Mahjoub Areej, Cihlarova Zuzana, Tétreault Martine, MacNeil Lauren, Sondheimer Neal, Caldecott Keith W, Hanzlikova Hana, Yoon Grace
Division of Neurology (A.M., G.Y.), Department of Paediatrics, University of Toronto, The Hospital for Sick Children, Canada; Department of Genome Dynamics (Z.C., K.W.C., H.H.), Institute of Molecular Genetics of the Czech Academy of Sciences, Prague, Czech Republic; Faculty of Science (Z.C.), Charles University in Prague, Czech Republic; Department of Neuroscience (M.T.), Université de Montréal, CHUM, Montréal, Québec, Canada; Department of Paediatric Laboratory Medicine (L.M.), Hospital for Sick Children; Department of Lab Medicine and Pathobiology (L.M.), University of Toronto, Ontario, Canada; Program in Genetics and Genome Biology (N.S.), SickKids Research Institute, Toronto, Ontario, Canada; Division of Clinical and Metabolic Genetics (N.S., G.Y.), Department of Paediatrics, University of Toronto, The Hospital for Sick Children, Toronto, Canada; and Genome Damage and Stability Centre (K.W.C., H.H.), School of Life Sciences, University of Sussex, Falmer, Brighton, UK.
Neurol Genet. 2019 Sep 4;5(5):e359. doi: 10.1212/NXG.0000000000000359. eCollection 2019 Oct.
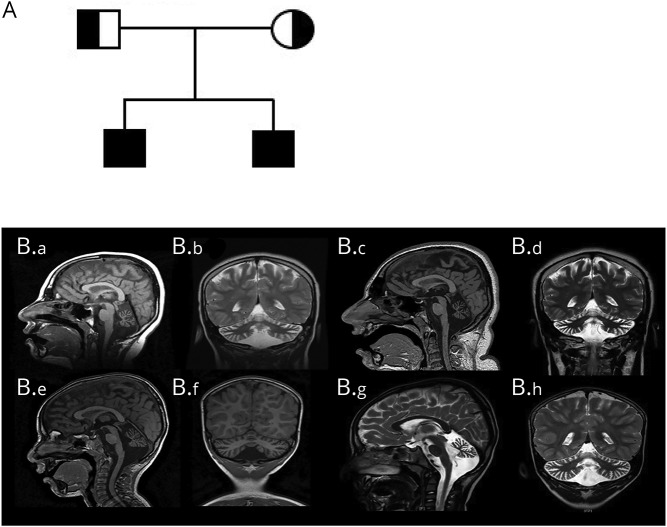
To investigate the pathogenicity of a novel homozygous variant in 2 siblings with nonprogressive cerebellar ataxia (NPCA) through functional studies on primary and immortalized patient cell lines.
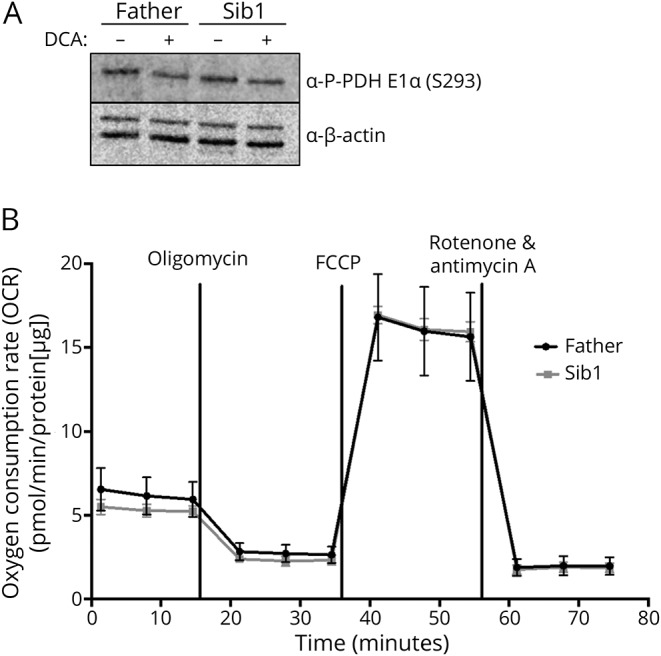
BRAT1 protein levels and ataxia-telangiectasia mutated (ATM) kinase activity in patient-derived and control cell lines were assessed by Western blotting. The impact of the novel variants on mitochondrial function was also assessed, by comparing patient and control cell lines for rates of oxygen consumption and for phosphorylation (S293) of the E1⍺ subunit of pyruvate dehydrogenase (PDH).
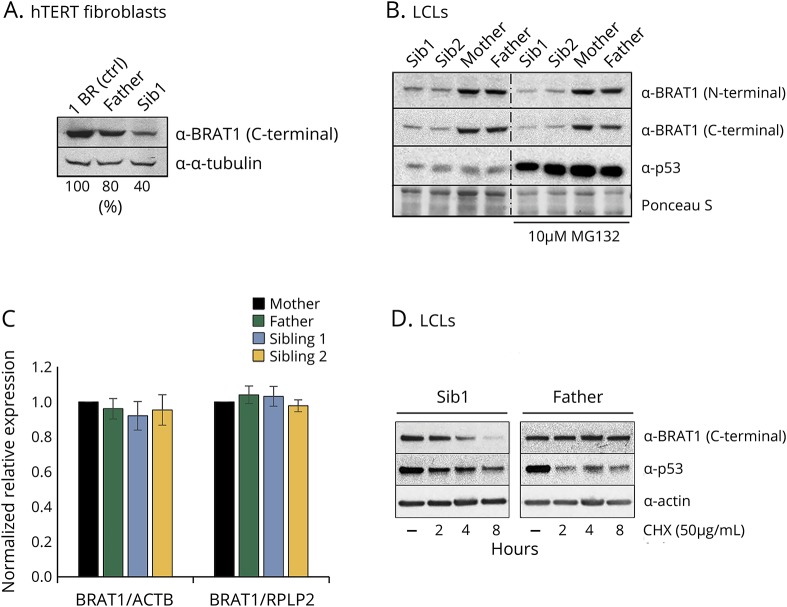
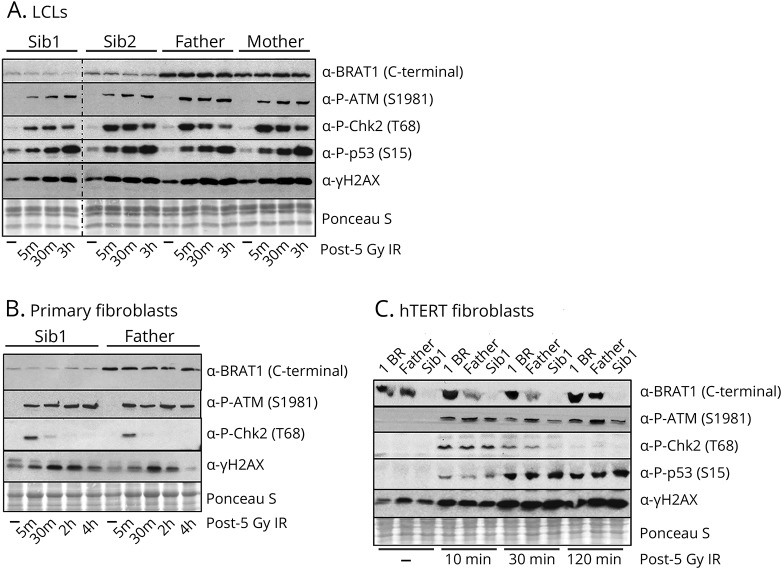
Two male siblings with NPCA, mild intellectual disability, and isolated cerebellar atrophy were found to be homozygous for a c.185T>A (p.Val62Glu) variant in by whole exome sequencing. Western blotting revealed markedly decreased BRAT1 protein levels in lymphocytes and/or fibroblast cells from both affected siblings compared to control cell lines. There were no differences between the patient and control cells in ATM kinase activation, following ionizing radiation. Mitochondrial studies were initially suggestive of a defect in regulation of PDH activity, but there was no evidence of increased phosphorylation of the E1⍺ subunit of the PDH complex. Measurement of oxygen consumption rates similarly failed to identify differences between patient and control cells.
Biallelic pathogenic variants in can be associated with NPCA, a phenotype considerably milder than previously reported. Surprisingly, despite the molecular role currently proposed for BRAT1 in ATM regulation, this disorder is unlikely to result from defective ATM kinase or mitochondrial dysfunction.
通过对原代和永生化患者细胞系进行功能研究,调查一种新的纯合变异体在2例非进行性小脑共济失调(NPCA)患者中的致病性。
通过蛋白质免疫印迹法评估患者来源和对照细胞系中BRAT1蛋白水平和共济失调毛细血管扩张症突变(ATM)激酶活性。还通过比较患者和对照细胞系的氧气消耗率以及丙酮酸脱氢酶(PDH)E1α亚基的磷酸化(S293)情况,评估该新变异体对线粒体功能的影响。
通过全外显子组测序发现,2例患有NPCA、轻度智力障碍和孤立性小脑萎缩的男性同胞在BRAT1基因上存在c.185T>A(p.Val62Glu)变异体的纯合情况。蛋白质免疫印迹法显示,与对照细胞系相比,两名受影响同胞的淋巴细胞和/或成纤维细胞中BRAT1蛋白水平明显降低。在电离辐射后,患者和对照细胞的ATM激酶激活情况没有差异。线粒体研究最初提示PDH活性调节存在缺陷,但没有证据表明PDH复合物的E1α亚基磷酸化增加。氧气消耗率的测量同样未能发现患者和对照细胞之间的差异。
BRAT1基因的双等位基因致病性变异体可能与NPCA相关,该表型比先前报道的要温和得多。令人惊讶的是,尽管目前认为BRAT1在ATM调节中具有分子作用,但这种疾病不太可能由ATM激酶缺陷或线粒体功能障碍引起。