School of Biotechnology, Dublin City University, Dublin, Ireland.
School of Medicine, University of, St. Andrews, UK.
Sci Rep. 2019 Nov 22;9(1):17394. doi: 10.1038/s41598-019-54004-5.
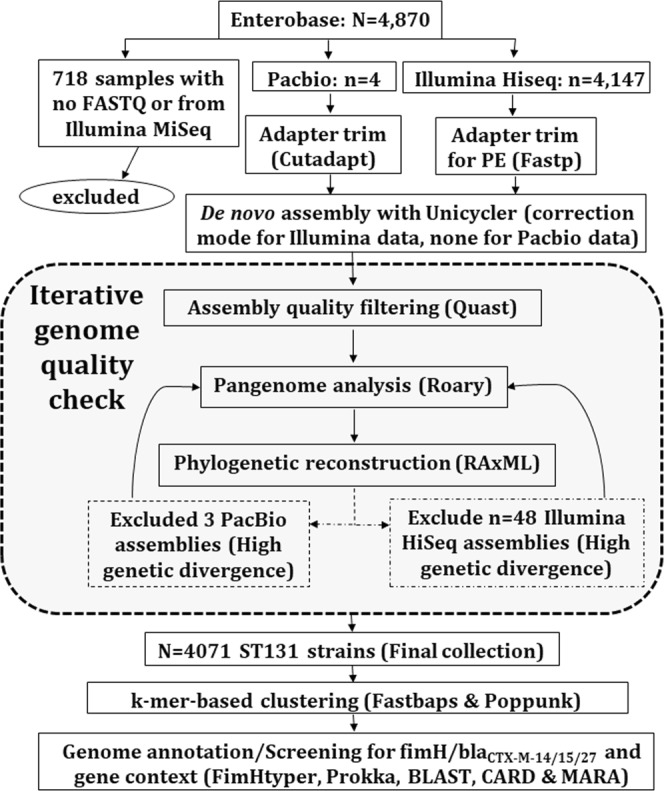
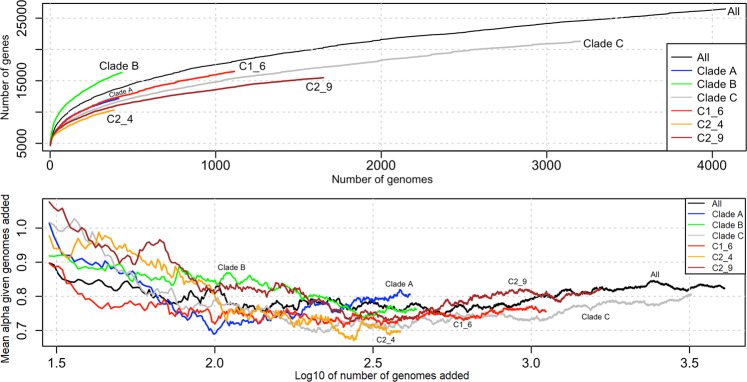
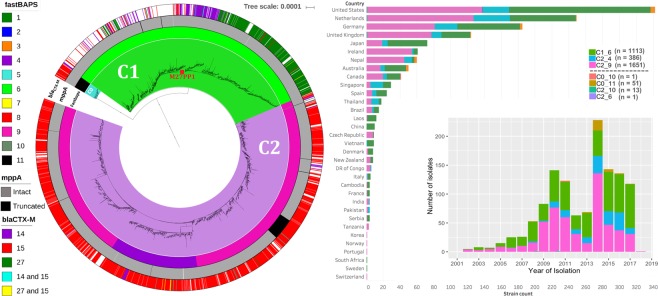
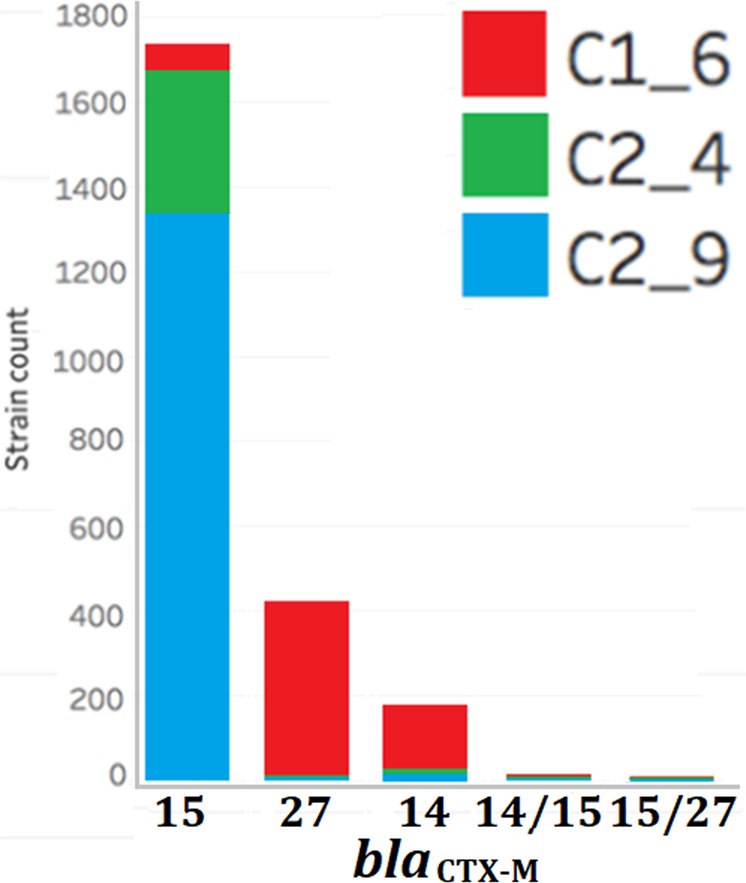
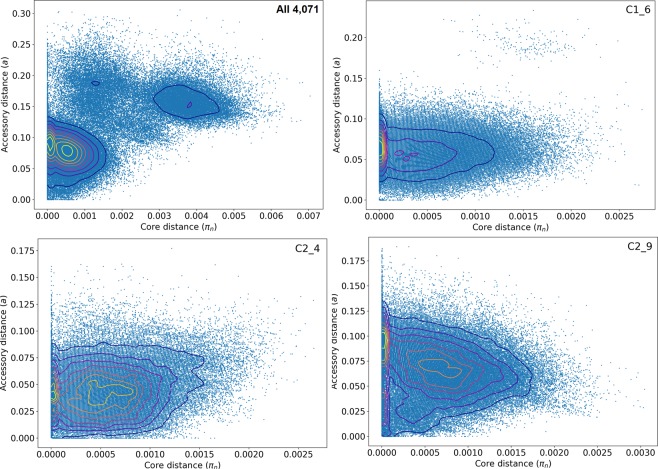
Escherichia coli ST131 is a major cause of infection with extensive antimicrobial resistance (AMR) facilitated by widespread beta-lactam antibiotic use. This drug pressure has driven extended-spectrum beta-lactamase (ESBL) gene acquisition and evolution in pathogens, so a clearer resolution of ST131's origin, adaptation and spread is essential. E. coli ST131's ESBL genes are typically embedded in mobile genetic elements (MGEs) that aid transfer to new plasmid or chromosomal locations, which are mobilised further by plasmid conjugation and recombination, resulting in a flexible ESBL, MGE and plasmid composition with a conserved core genome. We used population genomics to trace the evolution of AMR in ST131 more precisely by extracting all available high-quality Illumina HiSeq read libraries to investigate 4,071 globally-sourced genomes, the largest ST131 collection examined so far. We applied rigorous quality-control, genome de novo assembly and ESBL gene screening to resolve ST131's population structure across three genetically distinct Clades (A, B, C) and abundant subclades from the dominant Clade C. We reconstructed their evolutionary relationships across the core and accessory genomes using published reference genomes, long read assemblies and k-mer-based methods to contextualise pangenome diversity. The three main C subclades have co-circulated globally at relatively stable frequencies over time, suggesting attaining an equilibrium after their origin and initial rapid spread. This contrasted with their ESBL genes, which had stronger patterns across time, geography and subclade, and were located at distinct locations across the chromosomes and plasmids between isolates. Within the three C subclades, the core and accessory genome diversity levels were not correlated due to plasmid and MGE activity, unlike patterns between the three main clades, A, B and C. This population genomic study highlights the dynamic nature of the accessory genomes in ST131, suggesting that surveillance should anticipate genetically variable outbreaks with broader antibiotic resistance levels. Our findings emphasise the potential of evolutionary pangenomics to improve our understanding of AMR gene transfer, adaptation and transmission to discover accessory genome changes linked to novel subtypes.
大肠杆菌 ST131 是一种主要的感染源,其具有广泛的抗生素耐药性 (AMR),这是由于广泛使用β-内酰胺类抗生素所致。这种药物压力推动了病原体中扩展谱β-内酰胺酶 (ESBL) 基因的获得和进化,因此更清楚地了解 ST131 的起源、适应和传播至关重要。大肠杆菌 ST131 的 ESBL 基因通常嵌入在移动遗传元件 (MGEs) 中,这些元件有助于将基因转移到新的质粒或染色体位置,然后通过质粒接合和重组进一步移动,从而产生具有保守核心基因组的灵活 ESBL、MGE 和质粒组成。我们使用群体基因组学更精确地追踪 ST131 中 AMR 的进化,通过提取所有可用的高质量 Illumina HiSeq 读取文库来研究 4071 个全球来源的基因组,这是迄今为止检查的最大的 ST131 集合。我们应用严格的质量控制、基因组从头组装和 ESBL 基因筛选来解决 ST131 在三个遗传上不同的进化枝 (A、B、C) 和占主导地位的 C 进化枝中的大量亚进化枝中的种群结构。我们使用已发表的参考基因组、长读长组装和基于 k-mer 的方法,通过上下文化泛基因组多样性,在核心和辅助基因组上重建了它们的进化关系。三个主要的 C 亚进化枝在全球范围内相对稳定的频率下共同循环,这表明在起源和最初的快速传播后达到了平衡。这与它们的 ESBL 基因形成对比,这些基因在时间、地理和亚进化枝上具有更强的模式,并且位于分离株之间的染色体和质粒上的不同位置。在三个 C 亚进化枝中,核心和辅助基因组的多样性水平由于质粒和 MGE 的活性而没有相关性,这与三个主要进化枝 A、B 和 C 之间的模式不同。这项群体基因组研究强调了 ST131 辅助基因组的动态性质,表明监测应该预测具有更广泛抗生素耐药水平的基因变异爆发。我们的研究结果强调了进化泛基因组学在提高我们对 AMR 基因转移、适应和传播的理解方面的潜力,以发现与新型亚型相关的辅助基因组变化。