Signal Processing Laboratory, School of Engineering and Built Environment, Griffith University, Brisbane, QLD, 4111, Australia.
Institute for Glycomics and School of Information and Communication Technology, Griffith University, Parklands Dr., Southport, QLD, 4222, Australia.
Nat Commun. 2019 Nov 27;10(1):5407. doi: 10.1038/s41467-019-13395-9.
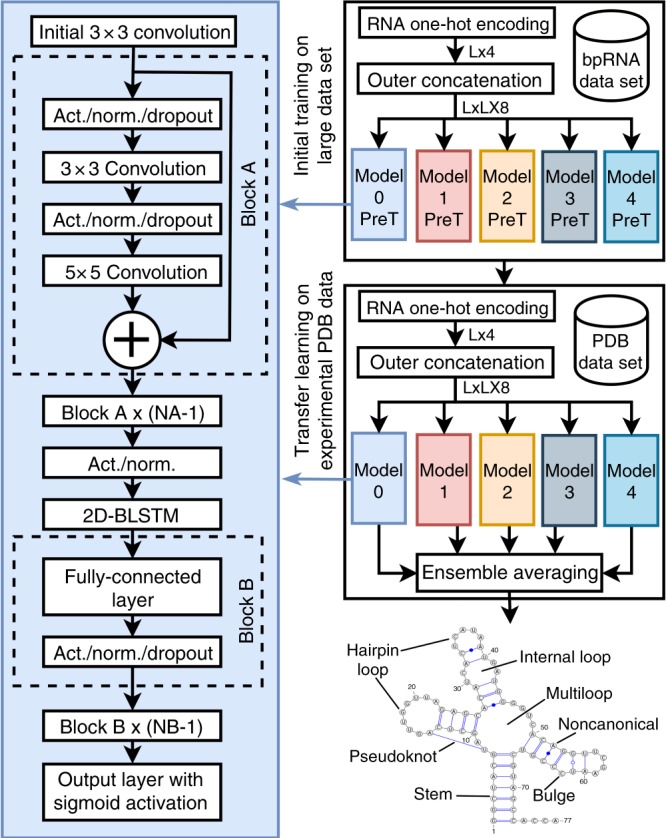
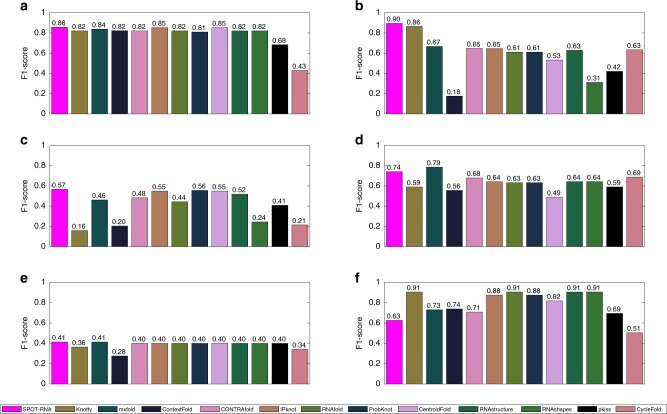
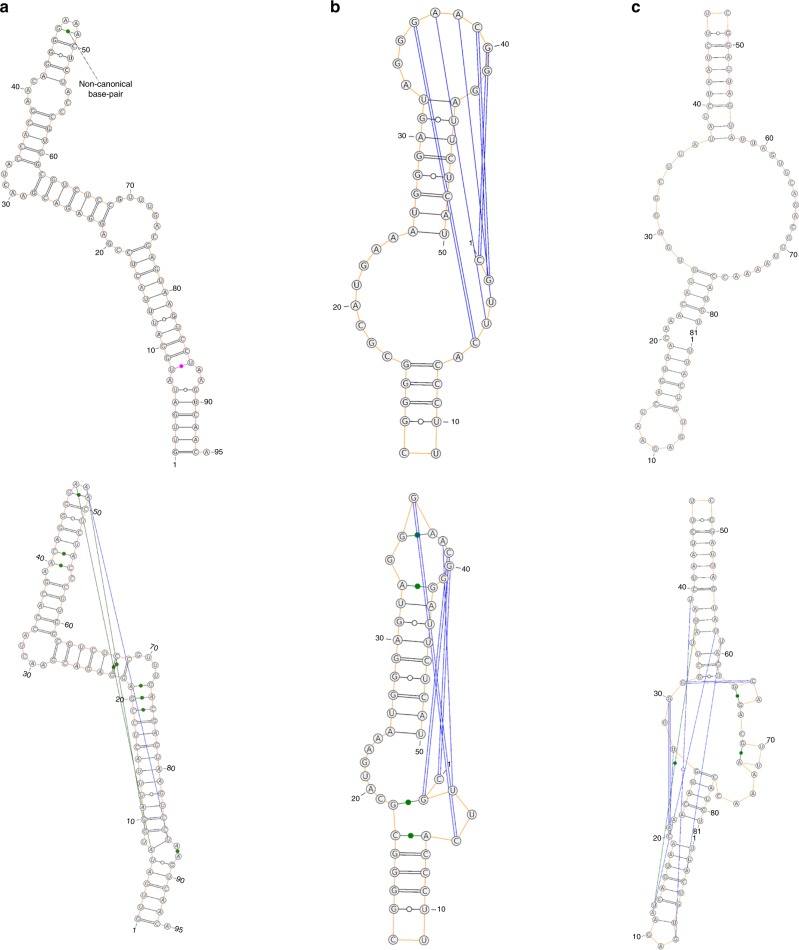
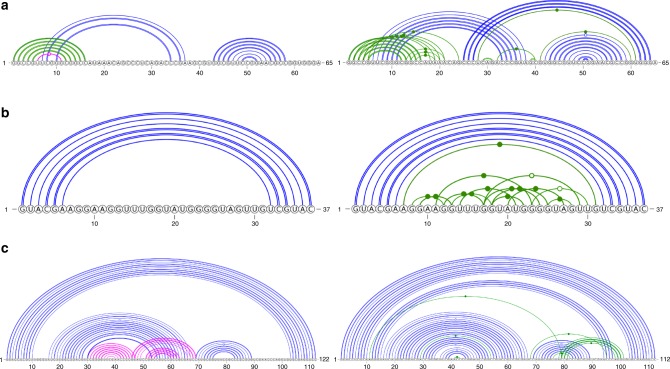
The majority of our human genome transcribes into noncoding RNAs with unknown structures and functions. Obtaining functional clues for noncoding RNAs requires accurate base-pairing or secondary-structure prediction. However, the performance of such predictions by current folding-based algorithms has been stagnated for more than a decade. Here, we propose the use of deep contextual learning for base-pair prediction including those noncanonical and non-nested (pseudoknot) base pairs stabilized by tertiary interactions. Since only [Formula: see text]250 nonredundant, high-resolution RNA structures are available for model training, we utilize transfer learning from a model initially trained with a recent high-quality bpRNA dataset of [Formula: see text]10,000 nonredundant RNAs made available through comparative analysis. The resulting method achieves large, statistically significant improvement in predicting all base pairs, noncanonical and non-nested base pairs in particular. The proposed method (SPOT-RNA), with a freely available server and standalone software, should be useful for improving RNA structure modeling, sequence alignment, and functional annotations.
我们人类基因组的大部分转录为具有未知结构和功能的非编码 RNA。获得非编码 RNA 的功能线索需要准确的碱基配对或二级结构预测。然而,目前基于折叠的算法在这些预测方面的性能已经停滞了十多年。在这里,我们提出使用深度上下文学习进行碱基对预测,包括那些由三级相互作用稳定的非规范和非嵌套(假结)碱基对。由于仅可用于模型训练的 [Formula: see text]250 个非冗余、高分辨率 RNA 结构,我们利用从最初使用最近高质量 bpRNA 数据集训练的模型进行迁移学习,该数据集通过比较分析获得了 [Formula: see text]10,000 个非冗余 RNA。所提出的方法在预测所有碱基对、特别是非规范和非嵌套碱基对方面取得了显著的、具有统计学意义的改进。该方法(SPOT-RNA)具有免费的服务器和独立的软件,应该有助于改进 RNA 结构建模、序列比对和功能注释。