Information and Computational Sciences, James Hutton Institute, Invergowrie, Dundee, DD2 5DA, UK.
Biomathematics and Statistics Scotland, Aberdeen, AB25 2ZD, UK.
BMC Genomics. 2019 Dec 11;20(1):968. doi: 10.1186/s12864-019-6243-7.
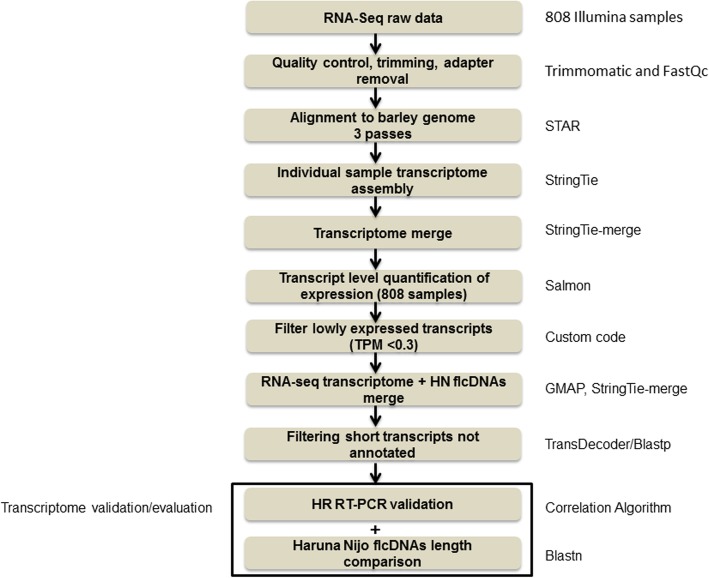
The time required to analyse RNA-seq data varies considerably, due to discrete steps for computational assembly, quantification of gene expression and splicing analysis. Recent fast non-alignment tools such as Kallisto and Salmon overcome these problems, but these tools require a high quality, comprehensive reference transcripts dataset (RTD), which are rarely available in plants.
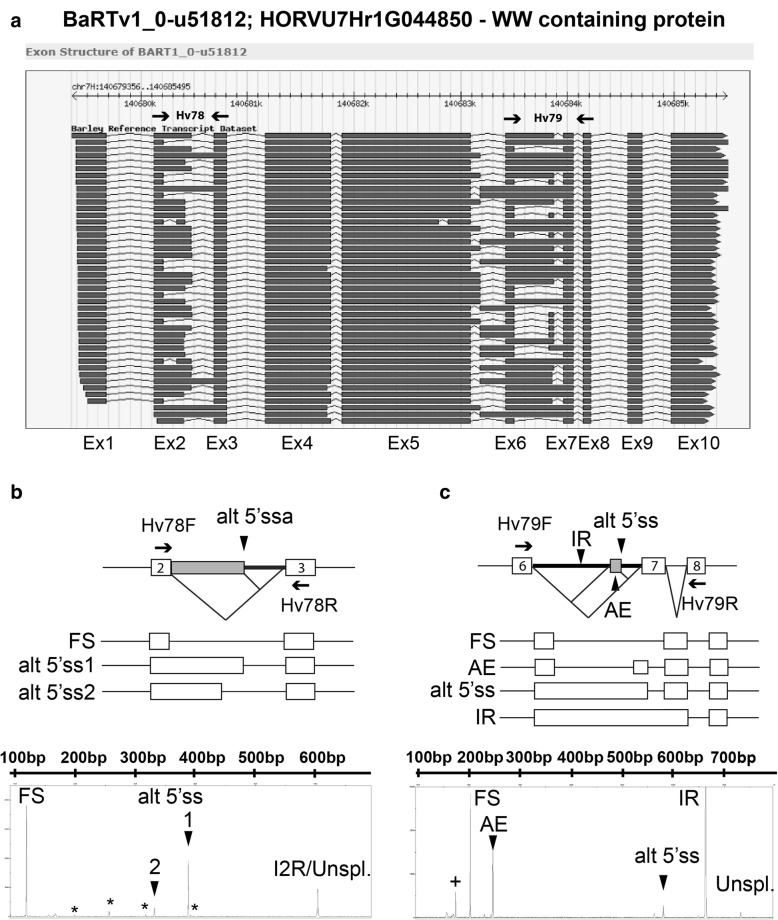
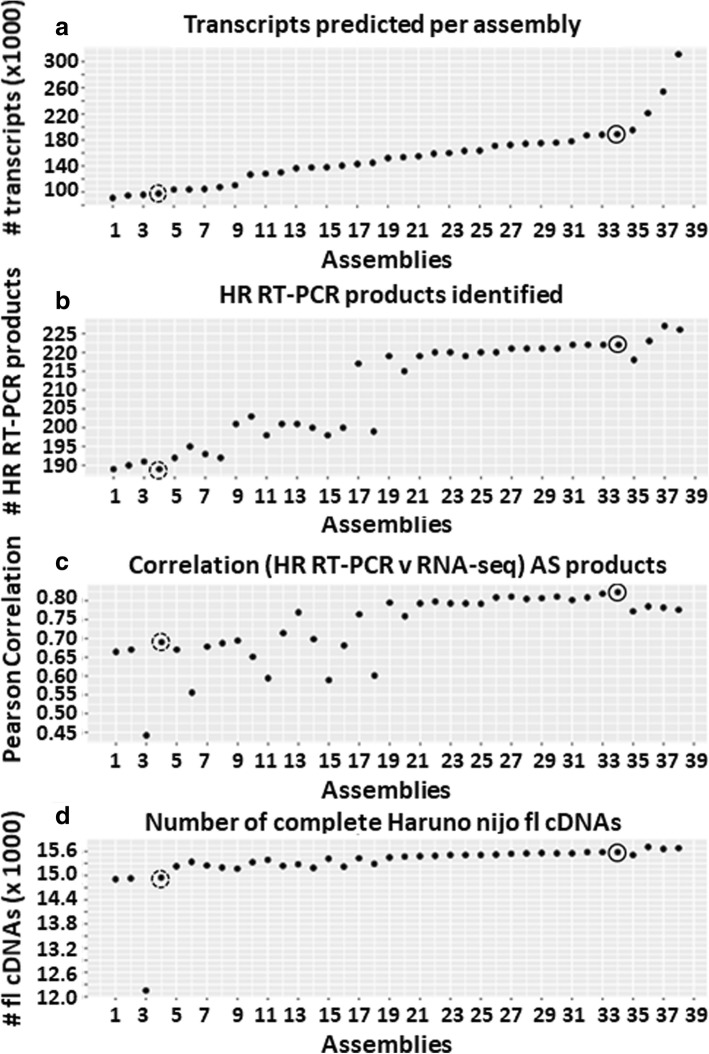
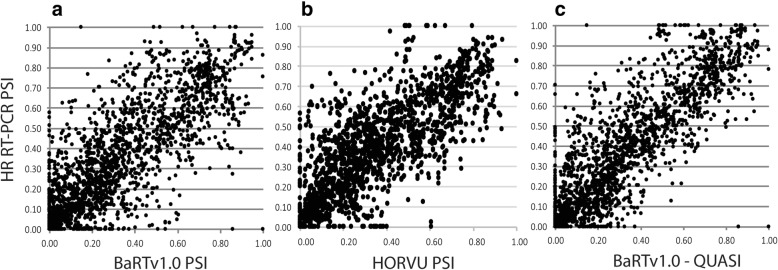
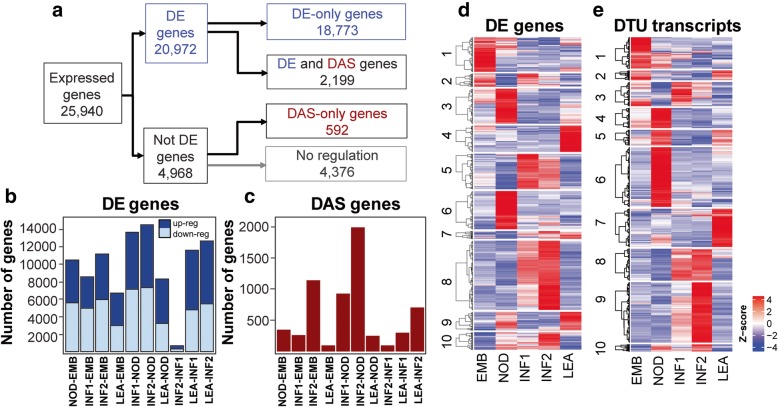
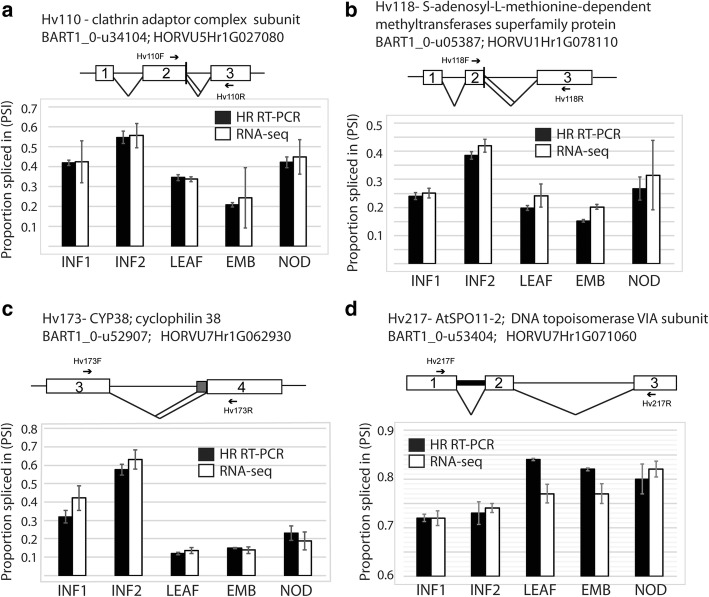
A high-quality, non-redundant barley gene RTD and database (Barley Reference Transcripts - BaRTv1.0) has been generated. BaRTv1.0, was constructed from a range of tissues, cultivars and abiotic treatments and transcripts assembled and aligned to the barley cv. Morex reference genome (Mascher et al. Nature; 544: 427-433, 2017). Full-length cDNAs from the barley variety Haruna nijo (Matsumoto et al. Plant Physiol; 156: 20-28, 2011) determined transcript coverage, and high-resolution RT-PCR validated alternatively spliced (AS) transcripts of 86 genes in five different organs and tissue. These methods were used as benchmarks to select an optimal barley RTD. BaRTv1.0-Quantification of Alternatively Spliced Isoforms (QUASI) was also made to overcome inaccurate quantification due to variation in 5' and 3' UTR ends of transcripts. BaRTv1.0-QUASI was used for accurate transcript quantification of RNA-seq data of five barley organs/tissues. This analysis identified 20,972 significant differentially expressed genes, 2791 differentially alternatively spliced genes and 2768 transcripts with differential transcript usage.
A high confidence barley reference transcript dataset consisting of 60,444 genes with 177,240 transcripts has been generated. Compared to current barley transcripts, BaRTv1.0 transcripts are generally longer, have less fragmentation and improved gene models that are well supported by splice junction reads. Precise transcript quantification using BaRTv1.0 allows routine analysis of gene expression and AS.
由于计算组装、基因表达定量和剪接分析的离散步骤,分析 RNA-seq 数据所需的时间差异很大。最近的快速非对齐工具,如 Kallisto 和 Salmon,克服了这些问题,但这些工具需要一个高质量、全面的参考转录本数据集(RTD),而这在植物中很少见。
生成了一个高质量、无冗余的大麦基因 RTD 和数据库(大麦参考转录本-BaRTv1.0)。BaRTv1.0 是由一系列组织、品种和非生物处理的转录本组装和比对到大麦 cv. Morex 参考基因组(Mascher 等人,《自然》;544:427-433,2017)构建的。来自大麦品种 Haruna nijo 的全长 cDNA(Matsumoto 等人,《植物生理学》;156:20-28,2011)确定了转录本覆盖度,高分辨率 RT-PCR 验证了 5 种不同器官和组织中 86 个基因的可变剪接(AS)转录本。这些方法被用作选择最佳大麦 RTD 的基准。还制作了 BaRTv1.0-可变剪接异构体定量(QUASI),以克服由于转录本 5'和 3'UTR 末端的变化导致的不准确定量。BaRTv1.0-QUASI 用于五个大麦器官/组织的 RNA-seq 数据的准确转录本定量。这项分析确定了 20972 个显著差异表达基因、2791 个差异可变剪接基因和 2768 个具有差异转录本使用的转录本。
生成了一个包含 60444 个基因和 177240 个转录本的高可信度大麦参考转录本数据集。与当前的大麦转录本相比,BaRTv1.0 转录本通常更长、碎片化更少,并且基因模型得到了剪接连接读取的良好支持。使用 BaRTv1.0 进行精确的转录本定量允许对基因表达和 AS 进行常规分析。