Wang Xiao, Su Guosheng, Hao Dan, Lund Mogens Sandø, Kadarmideen Haja N
1Quantitative Genomics, Bioinformatics and Computational Biology Group, Department of Applied Mathematics and Computer Science, Technical University of Denmark, Richard Peterson Plads, Building 324, 2800 Kongens Lyngby, Denmark.
2Center for Quantitative Genetics and Genomics, Department of Molecular Biology and Genetics, Aarhus University, 8830 Tjele, Denmark.
J Anim Sci Biotechnol. 2020 Jan 7;11:3. doi: 10.1186/s40104-019-0407-9. eCollection 2020.
Genotyping by sequencing (GBS) still has problems with missing genotypes. Imputation is important for using GBS for genomic predictions, especially for low depths, due to the large number of missing genotypes. Minor allele frequency (MAF) is widely used as a marker data editing criteria for genomic predictions. In this study, three imputation methods (Beagle, IMPUTE2 and FImpute software) based on four MAF editing criteria were investigated with regard to imputation accuracy of missing genotypes and accuracy of genomic predictions, based on simulated data of livestock population.
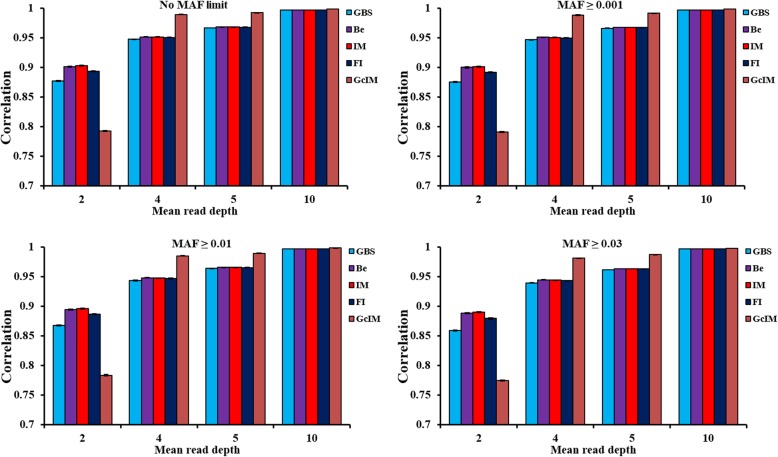
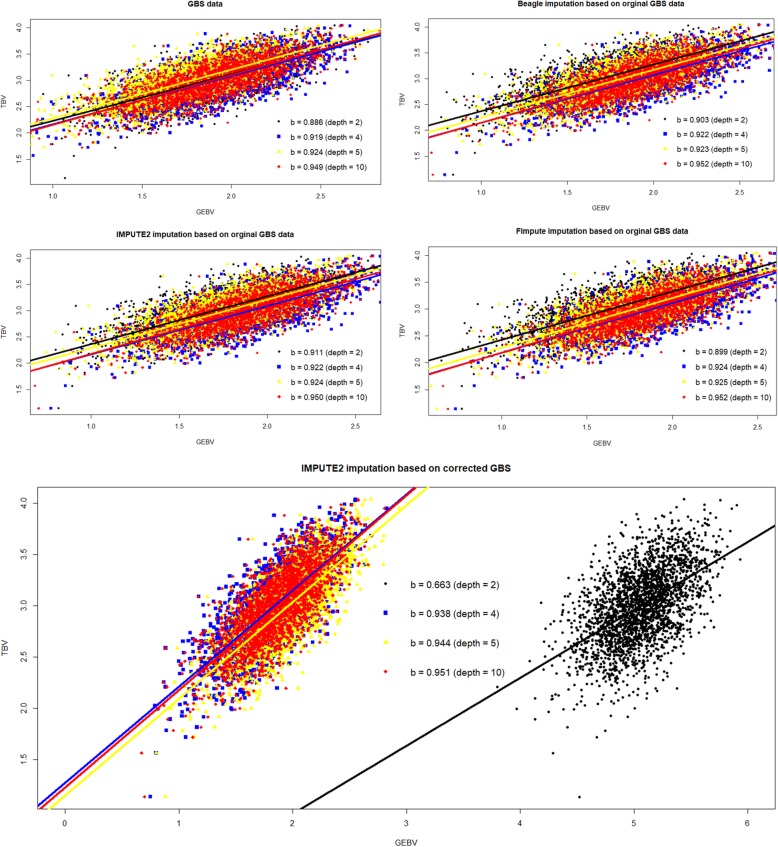
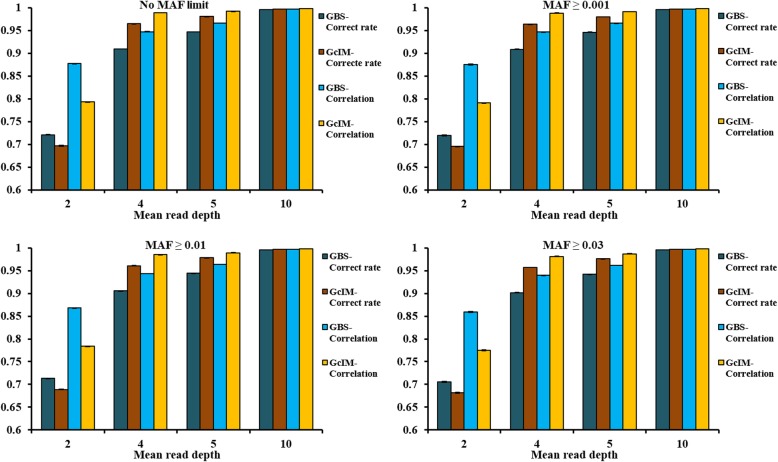
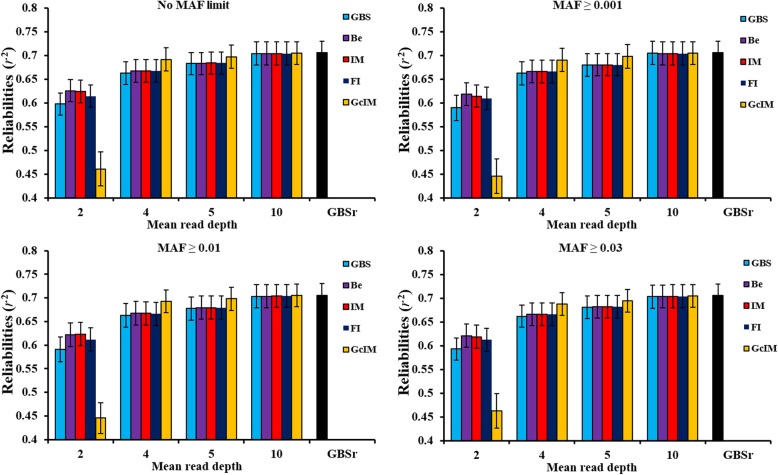
Four MAFs (no MAF limit, MAF ≥ 0.001, MAF ≥ 0.01 and MAF ≥ 0.03) were used for editing marker data before imputation. Beagle, IMPUTE2 and FImpute software were applied to impute the original GBS. Additionally, IMPUTE2 also imputed the expected genotype dosage after genotype correction (GcIM). The reliability of genomic predictions was calculated using GBS and imputed GBS data. The results showed that imputation accuracies were the same for the three imputation methods, except for the data of sequencing read depth (depth) = 2, where FImpute had a slightly lower imputation accuracy than Beagle and IMPUTE2. GcIM was observed to be the best for all of the imputations at depth = 4, 5 and 10, but the worst for depth = 2. For genomic prediction, retaining more SNPs with no MAF limit resulted in higher reliability. As the depth increased to 10, the prediction reliabilities approached those using true genotypes in the GBS loci. Beagle and IMPUTE2 had the largest increases in prediction reliability of 5 percentage points, and FImpute gained 3 percentage points at depth = 2. The best prediction was observed at depth = 4, 5 and 10 using GcIM, but the worst prediction was also observed using GcIM at depth = 2.
The current study showed that imputation accuracies were relatively low for GBS with low depths and high for GBS with high depths. Imputation resulted in larger gains in the reliability of genomic predictions for GBS with lower depths. These results suggest that the application of IMPUTE2, based on a corrected GBS (GcIM) to improve genomic predictions for higher depths, and FImpute software could be a good alternative for routine imputation.
测序基因分型(GBS)在基因型缺失方面仍存在问题。由于存在大量缺失基因型,插补对于将GBS用于基因组预测非常重要,尤其是对于低深度数据。小等位基因频率(MAF)被广泛用作基因组预测的标记数据编辑标准。在本研究中,基于家畜群体的模拟数据,研究了基于四种MAF编辑标准的三种插补方法(Beagle、IMPUTE2和FImpute软件)在缺失基因型插补准确性和基因组预测准确性方面的表现。
在插补前,使用四种MAF(无MAF限制、MAF≥0.001、MAF≥0.01和MAF≥0.03)编辑标记数据。应用Beagle、IMPUTE2和FImpute软件对原始GBS进行插补。此外,IMPUTE2还对基因型校正后的预期基因型剂量进行了插补(GcIM)。使用GBS和插补后的GBS数据计算基因组预测的可靠性。结果表明,除了测序读深度(深度)=2的数据外,三种插补方法的插补准确性相同,在该深度下,FImpute的插补准确性略低于Beagle和IMPUTE2。在深度=4、5和10时,观察到GcIM在所有插补中表现最佳,但在深度=2时表现最差。对于基因组预测,保留更多无MAF限制的单核苷酸多态性(SNP)会导致更高的可靠性。随着深度增加到10,预测可靠性接近使用GBS位点真实基因型时的可靠性。在深度=2时,Beagle和IMPUTE2的预测可靠性提高幅度最大,为5个百分点,FImpute提高了3个百分点。在深度=4、5和10时,使用GcIM观察到最佳预测,但在深度=2时使用GcIM也观察到最差预测。
当前研究表明,低深度GBS的插补准确性相对较低,高深度GBS的插补准确性较高。插补使低深度GBS的基因组预测可靠性有更大提高。这些结果表明,基于校正后的GBS(GcIM)应用IMPUTE2来改善高深度的基因组预测,FImpute软件可能是常规插补的一个不错选择。