Frischknecht Mirjam, Neuditschko Markus, Jagannathan Vidhya, Drögemüller Cord, Tetens Jens, Thaller Georg, Leeb Tosso, Rieder Stefan
Agroscope - Swiss National Stud Farm, 1580, Avenches, Switzerland.
Institute of Genetics, Vetsuisse Faculty, University of Bern, 3001, Bern, Switzerland.
Genet Sel Evol. 2014 Oct 1;46(1):63. doi: 10.1186/s12711-014-0063-7.
A cost-effective strategy to increase the density of available markers within a population is to sequence a small proportion of the population and impute whole-genome sequence data for the remaining population. Increased densities of typed markers are advantageous for genome-wide association studies (GWAS) and genomic predictions.
We obtained genotypes for 54 602 SNPs (single nucleotide polymorphisms) in 1077 Franches-Montagnes (FM) horses and Illumina paired-end whole-genome sequencing data for 30 FM horses and 14 Warmblood horses. After variant calling, the sequence-derived SNP genotypes (~13 million SNPs) were used for genotype imputation with the software programs Beagle, Impute2 and FImpute.
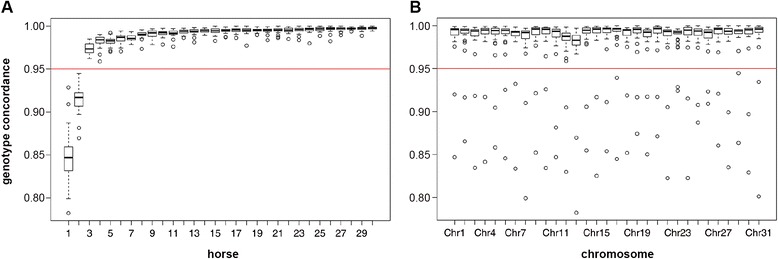
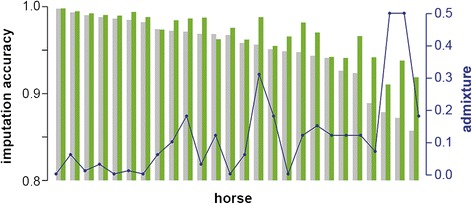
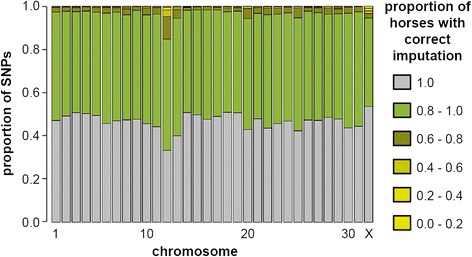
The mean imputation accuracy of FM horses using Impute2 was 92.0%. Imputation accuracy using Beagle and FImpute was 74.3% and 77.2%, respectively. In addition, for Impute2 we determined the imputation accuracy of all individual horses in the validation population, which ranged from 85.7% to 99.8%. The subsequent inclusion of Warmblood sequence data further increased the correlation between true and imputed genotypes for most horses, especially for horses with a high level of admixture. The final imputation accuracy of the horses ranged from 91.2% to 99.5%.
Using Impute2, the imputation accuracy was higher than 91% for all horses in the validation population, which indicates that direct imputation of 50k SNP-chip data to sequence level genotypes is feasible in the FM population. The individual imputation accuracy depended mainly on the applied software and the level of admixture.
在一个群体中增加可用标记密度的一种经济有效的策略是对一小部分群体进行测序,并为其余群体推算全基因组序列数据。增加分型标记的密度对全基因组关联研究(GWAS)和基因组预测是有利的。
我们获得了1077匹弗朗什-蒙塔涅(FM)马中54602个单核苷酸多态性(SNP)的基因型,以及30匹FM马和14匹温血马的Illumina双端全基因组测序数据。在进行变异检测后,将序列衍生的SNP基因型(约1300万个SNP)用于使用Beagle、Impute2和FImpute软件程序进行基因型推算。
使用Impute2对FM马进行推算的平均准确率为92.0%。使用Beagle和FImpute的推算准确率分别为74.3%和77.2%。此外,对于Impute2,我们确定了验证群体中所有个体马的推算准确率,范围为85.7%至99.8%。随后纳入温血马序列数据进一步提高了大多数马的真实基因型与推算基因型之间的相关性,特别是对于具有高度混合水平的马。这些马的最终推算准确率范围为从91.2%至99.5%。
使用Impute2,验证群体中所有马的推算准确率均高于91%,这表明在FM群体中将50k SNP芯片数据直接推算到序列水平基因型是可行的。个体推算准确率主要取决于所应用的软件和混合水平。