Emperador Sonia, Garrido-Pérez Nuria, Amezcua-Gil Javier, Gaudó Paula, Andrés-Sanz Julio Alberto, Yubero Delia, Fernández-Marmiesse Ana, O'Callaghan Maria M, Ortigoza-Escobar Juan D, Iriondo Marti, Ruiz-Pesini Eduardo, García-Cazorla Angels, Gil-Campos Mercedes, Artuch Rafael, Montoya Julio, Bayona-Bafaluy María Pilar
Departamento de Bioquímica, Biología Molecular y Celular, Universidad de Zaragoza, Zaragoza, Spain.
Instituto de Investigación Sanitaria de Aragón (IIS-Aragón), Zaragoza, Spain.
Front Genet. 2020 Jan 8;10:1300. doi: 10.3389/fgene.2019.01300. eCollection 2019.
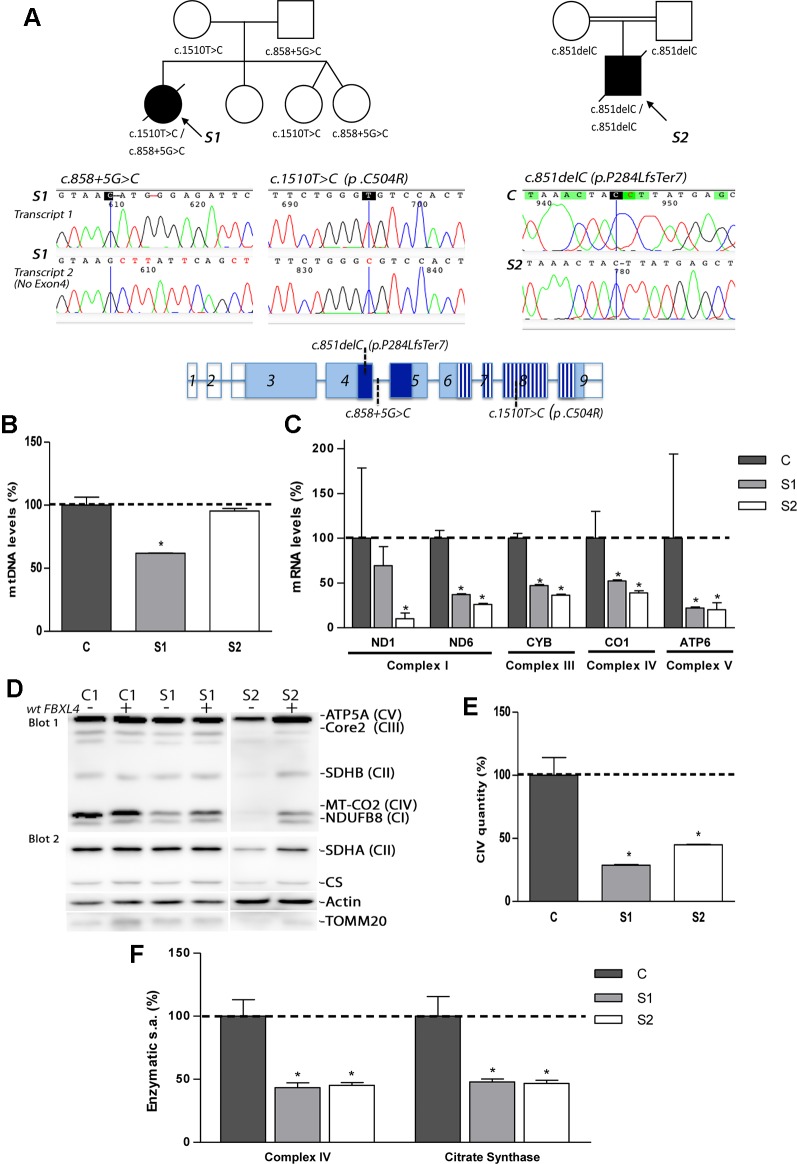
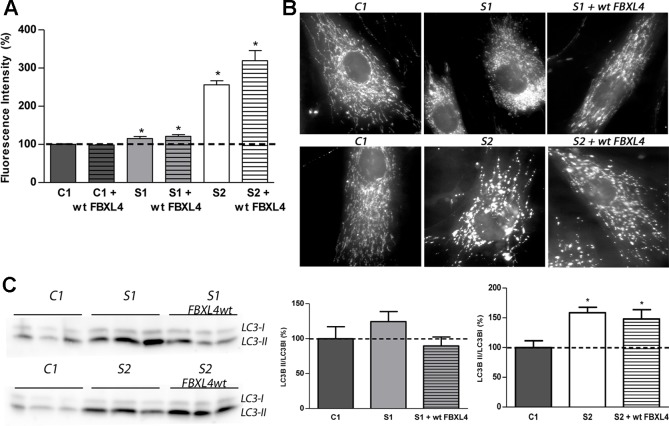
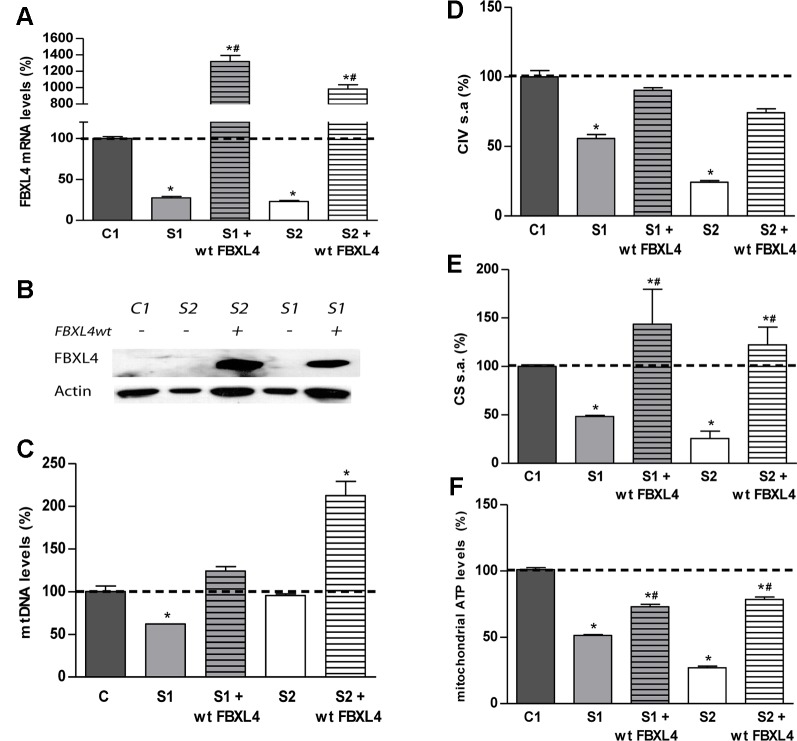
Encephalomyopathic mitochondrial DNA (mtDNA) depletion syndrome 13 (MTDPS13) is a rare genetic disorder caused by defects in F-box leucine-rich repeat protein 4 (FBXL4). Although FBXL4 is essential for the bioenergetic homeostasis of the cell, the precise role of the protein remains unknown. In this study, we report two cases of unrelated patients presenting in the neonatal period with hyperlactacidemia and generalized hypotonia. Severe mtDNA depletion was detected in muscle biopsy in both patients. Genetic analysis showed one patient as having in compound heterozygosis a splice site variant c.858+5G>C and a missense variant c.1510T>C (p.Cys504Arg) in . The second patient harbored a frameshift novel variant c.851delC (p.Pro284LeufsTer7) in homozygosis. To validate the pathogenicity of these variants, molecular and biochemical analyses were performed using skin-derived fibroblasts. We observed that the mtDNA depletion was less severe in fibroblasts than in muscle. Interestingly, the cells harboring a nonsense variant in homozygosis showed normal mtDNA copy number. Both patient fibroblasts, however, demonstrated reduced mitochondrial transcript quantity leading to diminished steady state levels of respiratory complex subunits, decreased respiratory complex IV (CIV) activity, and finally, low mitochondrial ATP levels. Both patients also revealed citrate synthase deficiency. Genetic complementation assays established that the deficient phenotype was rescued by the canonical version of , confirming the pathological nature of the variants. Further analysis of fibroblasts allowed to establish that increased mitochondrial mass, mitochondrial fragmentation, and augmented autophagy are associated with FBXL4 deficiency in cells, but are probably secondary to a primary metabolic defect affecting oxidative phosphorylation.
脑肌病性线粒体DNA(mtDNA)耗竭综合征13(MTDPS13)是一种由F-box富含亮氨酸重复蛋白4(FBXL4)缺陷引起的罕见遗传疾病。虽然FBXL4对细胞的生物能量稳态至关重要,但该蛋白的确切作用仍然未知。在本研究中,我们报告了两例无关患者,他们在新生儿期出现高乳酸血症和全身肌张力减退。两名患者的肌肉活检均检测到严重的mtDNA耗竭。基因分析显示,一名患者为复合杂合子,存在剪接位点变异c.858 + 5G>C和错义变异c.1510T>C(p.Cys504Arg)。第二名患者纯合子携带移码新变异c.851delC(p.Pro284LeufsTer7)。为了验证这些变异的致病性,使用皮肤来源的成纤维细胞进行了分子和生化分析。我们观察到,成纤维细胞中的mtDNA耗竭比肌肉中的要轻。有趣的是,纯合子携带无义变异的细胞显示出正常的mtDNA拷贝数。然而,两名患者的成纤维细胞均表现出线粒体转录物数量减少,导致呼吸复合体亚基的稳态水平降低、呼吸复合体IV(CIV)活性降低,最终线粒体ATP水平降低。两名患者还表现出柠檬酸合酶缺乏。基因互补试验证实,野生型可挽救缺陷表型,从而证实了这些变异的病理性质。对成纤维细胞的进一步分析表明,线粒体质量增加、线粒体碎片化和自噬增强与细胞中FBXL4缺乏有关,但可能继发于影响氧化磷酸化的原发性代谢缺陷。