Département de biochimie et génomique fonctionnelle, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke, QC J1E 4K8, Canada.
Département de microbiologie et d'infectiologie, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke, QC J1E 4K8, Canada.
Nucleic Acids Res. 2020 Mar 18;48(5):2271-2286. doi: 10.1093/nar/gkaa028.
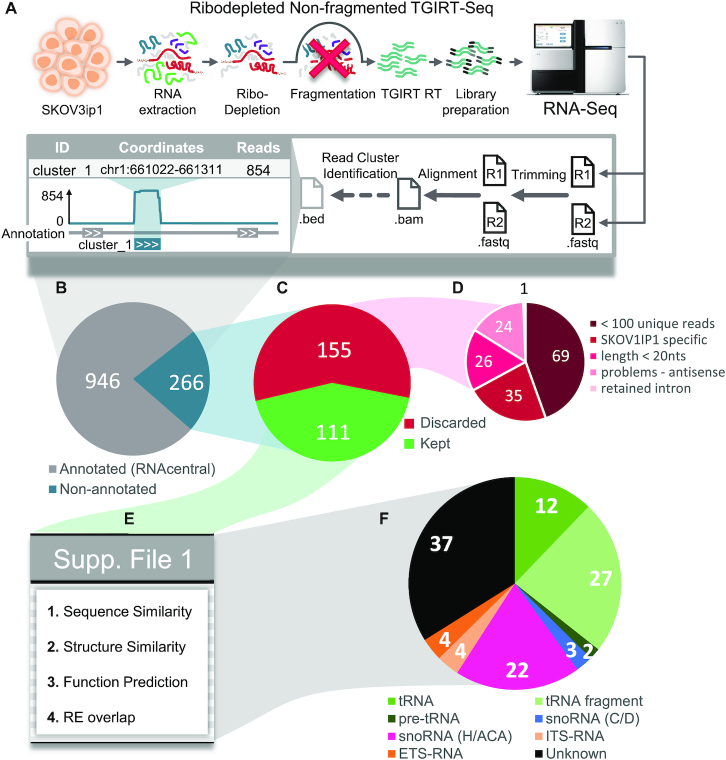
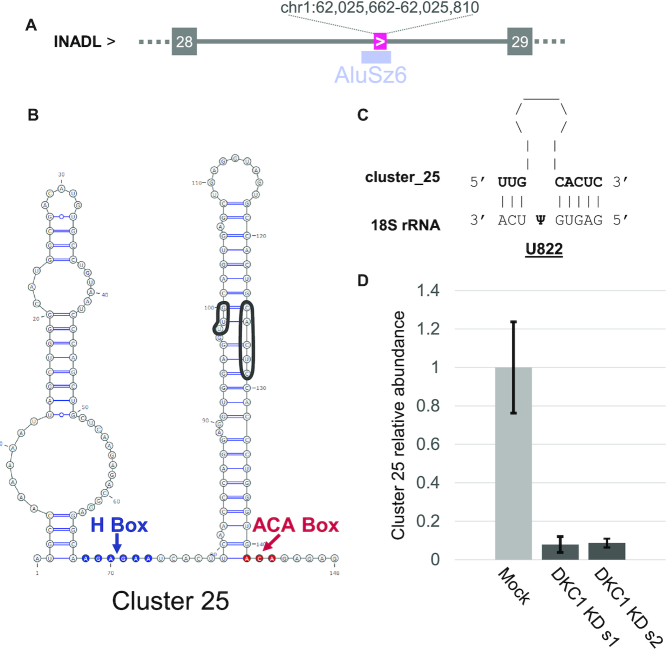
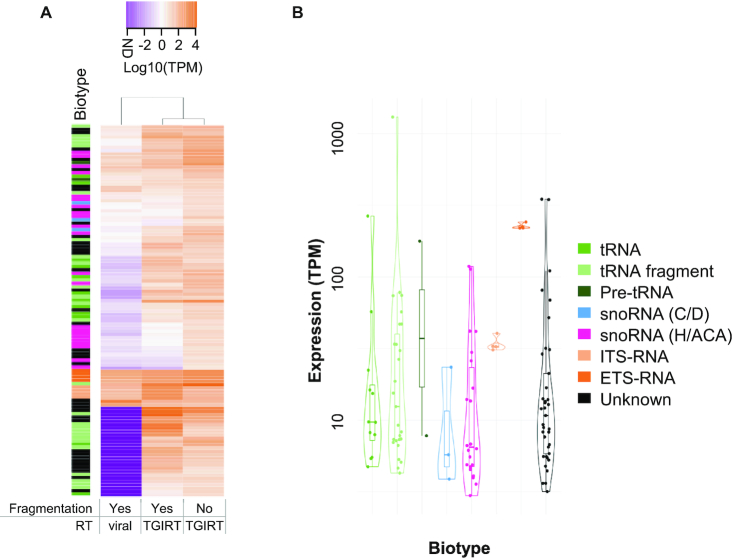
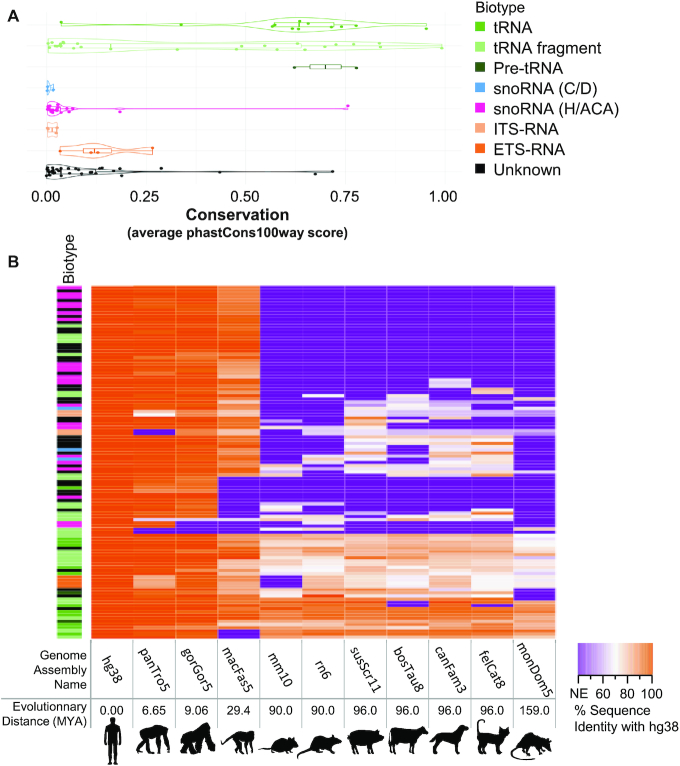
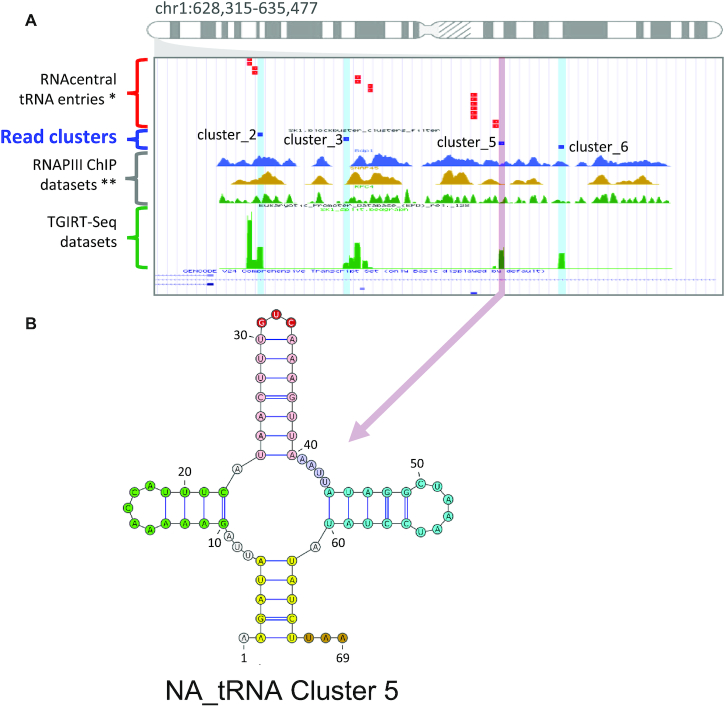
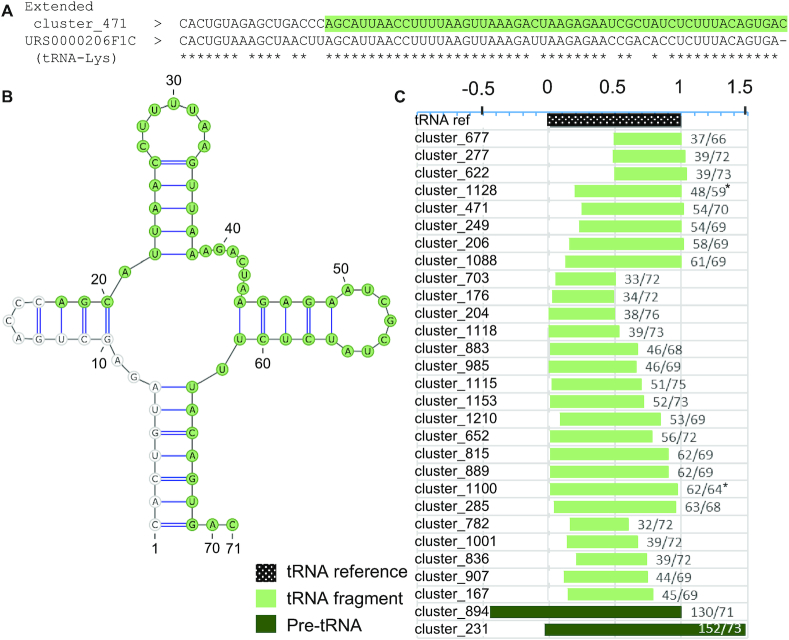
The study of RNA expression is the fastest growing area of genomic research. However, despite the dramatic increase in the number of sequenced transcriptomes, we still do not have accurate estimates of the number and expression levels of non-coding RNA genes. Non-coding transcripts are often overlooked due to incomplete genome annotation. In this study, we use annotation-independent detection of RNA reads generated using a reverse transcriptase with low structure bias to identify non-coding RNA. Transcripts between 20 and 500 nucleotides were filtered and crosschecked with non-coding RNA annotations revealing 111 non-annotated non-coding RNAs expressed in different cell lines and tissues. Inspecting the sequence and structural features of these transcripts indicated that 60% of these transcripts correspond to new snoRNA and tRNA-like genes. The identified genes exhibited features of their respective families in terms of structure, expression, conservation and response to depletion of interacting proteins. Together, our data reveal a new group of RNA that are difficult to detect using standard gene prediction and RNA sequencing techniques, suggesting that reliance on actual gene annotation and sequencing techniques distorts the perceived architecture of the human transcriptome.
RNA 表达研究是基因组研究中发展最快的领域。然而,尽管转录组测序数量急剧增加,但我们仍然无法准确估计非编码 RNA 基因的数量和表达水平。由于基因组注释不完整,非编码转录本经常被忽视。在这项研究中,我们使用反转录酶进行 RNA reads 的无注释检测,以识别非编码 RNA。筛选出 20 到 500 个核苷酸的转录本,并与非编码 RNA 注释进行交叉检查,结果显示在不同的细胞系和组织中表达了 111 个未注释的非编码 RNA。检查这些转录本的序列和结构特征表明,其中 60%的转录本对应于新的 snoRNA 和 tRNA 样基因。鉴定出的基因在结构、表达、保守性和对相互作用蛋白缺失的反应方面表现出各自家族的特征。总之,我们的数据揭示了一组新的 RNA,这些 RNA 很难用标准的基因预测和 RNA 测序技术检测到,这表明依赖于实际的基因注释和测序技术会扭曲人们对人类转录组结构的认识。