Al-Refaei Maha Ateeq, Makki Rania Marwan, Ali Hani Mohammed
Department of Biological Sciences, Faculty of Sciences, King Abdulaziz University, Jeddah, Saudi Arabia.
Heliyon. 2020 Jan 29;6(1):e03221. doi: 10.1016/j.heliyon.2020.e03221. eCollection 2020 Jan.
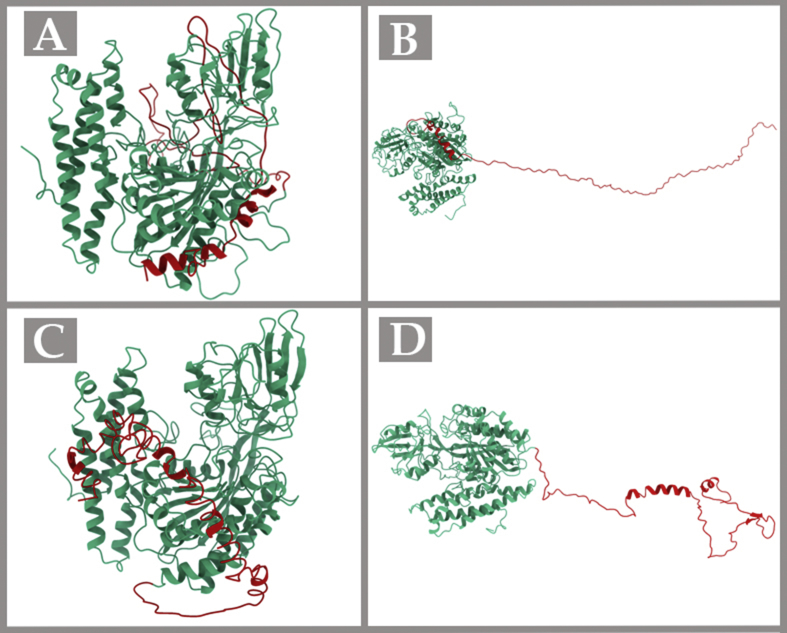
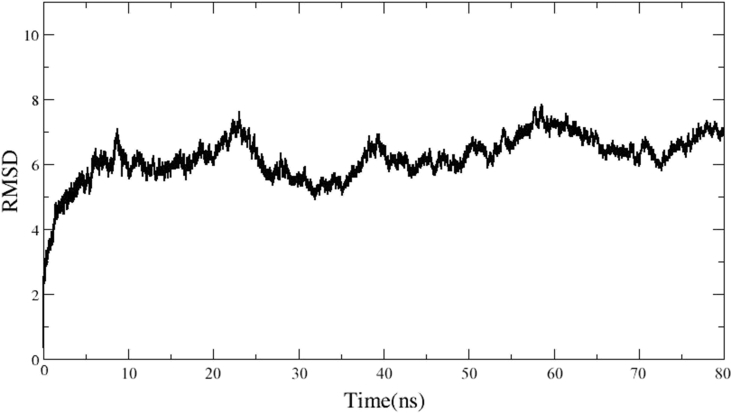
Transferrin receptor protein 1 (TfR1) is an important molecule in anti-cancer therapy. Targeted delivery of such therapeutic compounds improves their cellular uptake and circulation time, thereby enhancing therapeutic efficacy. Drug designing is therefore used to engineer molecules with structures that facilitate specific interactions. However, this process requires a thorough knowledge of all the interactions, including the three-dimensional (3D) and quaternary structures (QS) of the interacting molecules. Since structural information is available for only a part of the full TfR1 sequence, in the present study, we predicted the whole structure of TfR1 using homology modelling, docking, and molecular dynamics simulations. Homology modelling is used to generate 3D structures of TfR1 using MODELLER, I-TASSER, and RaptorX programs. Verify3D and Rampage server evaluated the quality of the resultant models. According to this evaluation, the model built by the RaptorX server and validated by Verify3D (compatibility: 83.82%) had the highest number of residues (95.5%) within the favoured regions of the Ramachandran plot, making it the most reliable 3D protein structure for TfR1 compared with others. The QS of TfR1 was built using HADDOCK and SymmDock docking software, and the results were evaluated by the ligand root mean square deviation (l-RMSD) value computed using the ProFit software. This showed that both HADDOCK and SymmDock gave acceptable results. However, the HADDOCK result was more stable and closest to the native complex structure with disulfide bonds. Therefore, the HADDOCK complex was further refined using both SymmRef and GalaxyRefineComplex until the medium l-RMSD rank was reached. This QS was successfully verified using nanoscale molecular dynamics (NAMD) energy minimization. This model could pave the way for further functional, structural, and therapeutic studies on TfR1.
转铁蛋白受体蛋白1(TfR1)是抗癌治疗中的一个重要分子。此类治疗性化合物的靶向递送可改善其细胞摄取和循环时间,从而提高治疗效果。因此,药物设计用于构建具有促进特定相互作用结构的分子。然而,这个过程需要全面了解所有相互作用,包括相互作用分子的三维(3D)结构和四级结构(QS)。由于仅获得了完整TfR1序列一部分的结构信息,在本研究中,我们使用同源建模、对接和分子动力学模拟预测了TfR1的整体结构。同源建模用于使用MODELLER、I-TASSER和RaptorX程序生成TfR1的3D结构。Verify3D和Rampage服务器评估所得模型的质量。根据该评估,由RaptorX服务器构建并经Verify3D验证(兼容性:83.82%)的模型在拉氏图的有利区域内具有最多数量的残基(95.5%),使其成为与其他模型相比最可靠的TfR1的3D蛋白质结构。TfR1的QS使用HADDOCK和SymmDock对接软件构建,结果通过使用ProFit软件计算的配体均方根偏差(l-RMSD)值进行评估。这表明HADDOCK和SymmDock均给出了可接受的结果。然而,HADDOCK的结果更稳定且最接近具有二硫键的天然复合物结构。因此,使用SymmRef和GalaxyRefineComplex对HADDOCK复合物进行进一步优化,直至达到中等l-RMSD排名。使用纳米尺度分子动力学(NAMD)能量最小化成功验证了该QS。该模型可为TfR1的进一步功能、结构和治疗研究铺平道路。