Department of Plant and Environmental Sciences, University of Copenhagen, Thorvaldsensvej 40, Frederiksberg C, 1871, Denmark.
Department of Environmental Science, Aarhus University, Frederiksborgvej 399, Roskilde, 4000, Denmark.
Gigascience. 2020 Feb 1;9(2). doi: 10.1093/gigascience/giaa008.
Metagenomic sequencing is a well-established tool in the modern biosciences. While it promises unparalleled insights into the genetic content of the biological samples studied, conclusions drawn are at risk from biases inherent to the DNA sequencing methods, including inaccurate abundance estimates as a function of genomic guanine-cytosine (GC) contents.
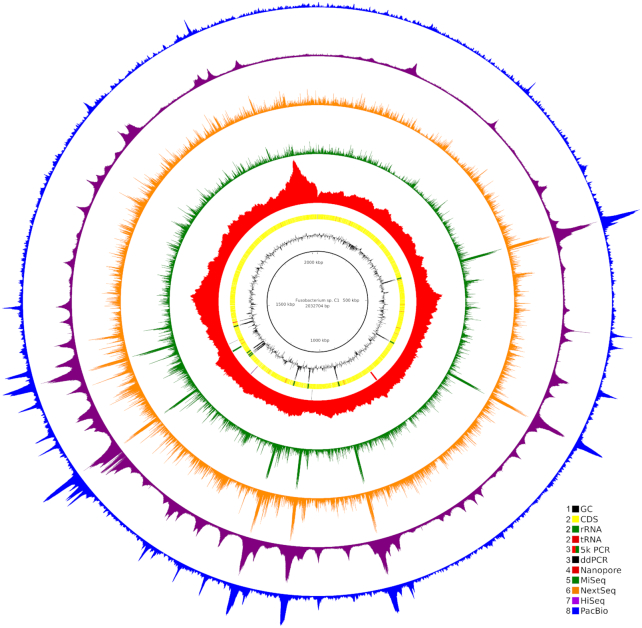
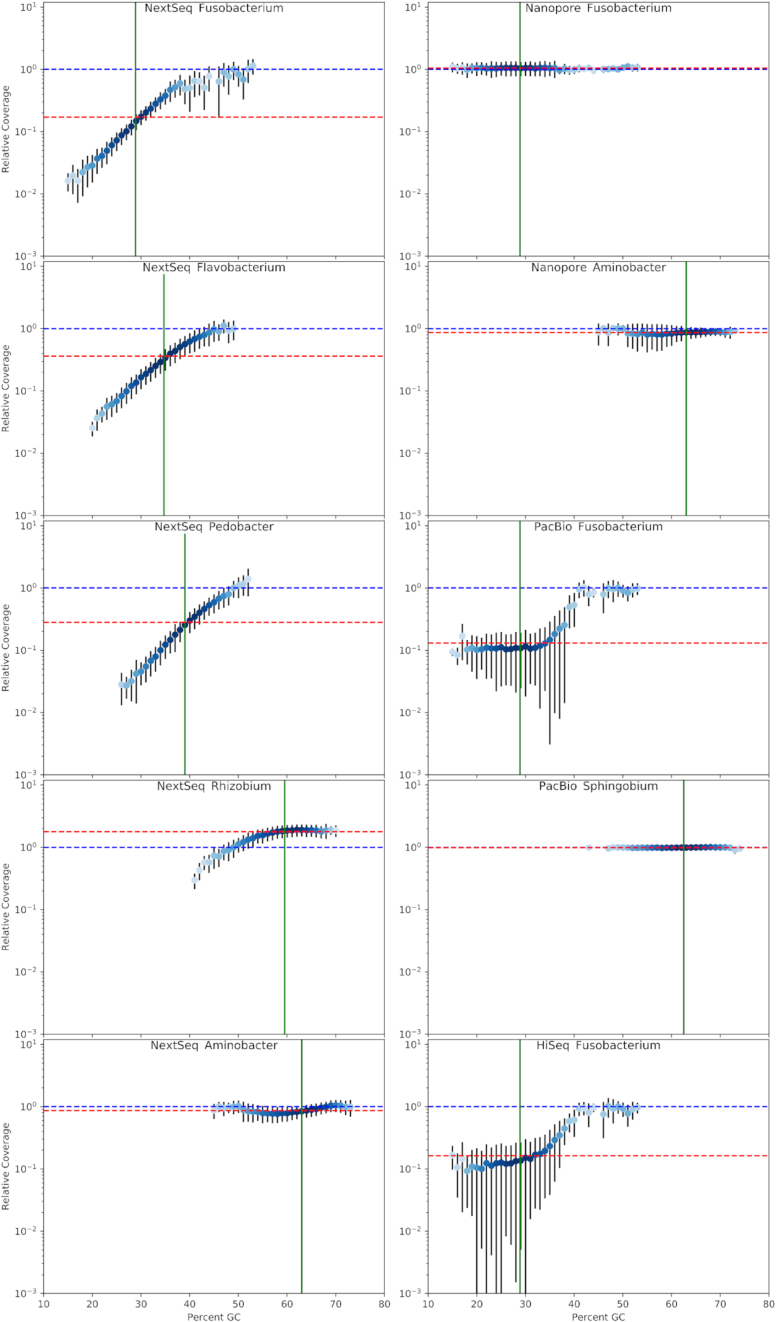
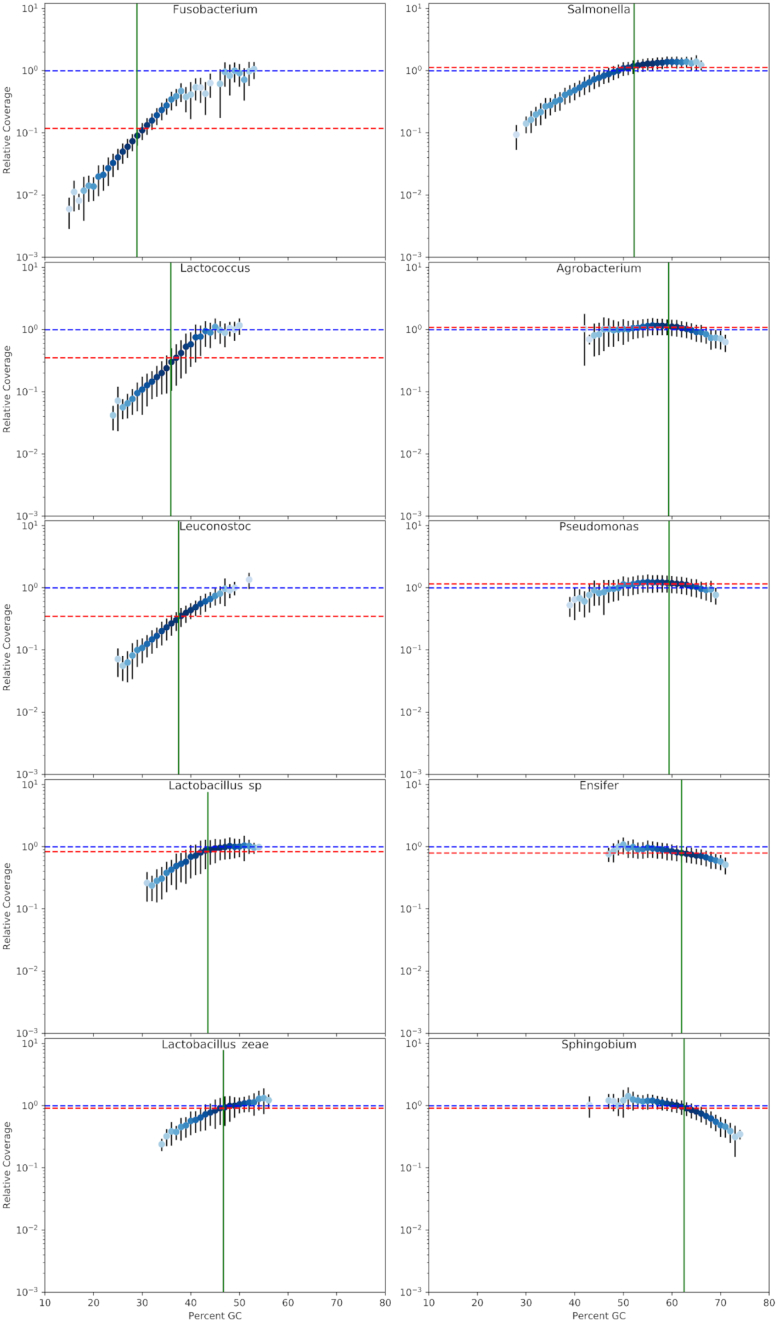
We explored such GC biases across many commonly used platforms in experiments sequencing multiple genomes (with mean GC contents ranging from 28.9% to 62.4%) and metagenomes. GC bias profiles varied among different library preparation protocols and sequencing platforms. We found that our workflows using MiSeq and NextSeq were hindered by major GC biases, with problems becoming increasingly severe outside the 45-65% GC range, leading to a falsely low coverage in GC-rich and especially GC-poor sequences, where genomic windows with 30% GC content had >10-fold less coverage than windows close to 50% GC content. We also showed that GC content correlates tightly with coverage biases. The PacBio and HiSeq platforms also evidenced similar profiles of GC biases to each other, which were distinct from those seen in the MiSeq and NextSeq workflows. The Oxford Nanopore workflow was not afflicted by GC bias.
These findings indicate potential sources of difficulty, arising from GC biases, in genome sequencing that could be pre-emptively addressed with methodological optimizations provided that the GC biases inherent to the relevant workflow are understood. Furthermore, it is recommended that a more critical approach be taken in quantitative abundance estimates in metagenomic studies. In the future, metagenomic studies should take steps to account for the effects of GC bias before drawing conclusions, or they should use a demonstrably unbiased workflow.
宏基因组测序是现代生物科学中一种成熟的工具。虽然它有望提供对所研究生物样本遗传内容的无与伦比的见解,但由于 DNA 测序方法固有的偏差,包括作为基因组鸟嘌呤-胞嘧啶(GC)含量函数的不准确丰度估计,得出的结论存在风险。
我们在多个常用平台上进行了实验,对多个基因组(GC 含量从 28.9%到 62.4%不等)和宏基因组进行了测序,以探索这种 GC 偏差。GC 偏差谱在不同的文库制备方案和测序平台之间有所不同。我们发现,我们使用 MiSeq 和 NextSeq 的工作流程受到了主要 GC 偏差的阻碍,在 45-65%GC 范围之外,问题变得越来越严重,导致 GC 丰富和特别是 GC 贫乏的序列覆盖度过低,其中 GC 含量为 30%的基因组窗口的覆盖度比接近 50%GC 含量的窗口低 10 倍以上。我们还表明,GC 含量与覆盖度偏差密切相关。PacBio 和 HiSeq 平台彼此之间也存在相似的 GC 偏差分布,与 MiSeq 和 NextSeq 工作流程中的偏差明显不同。Oxford Nanopore 工作流程不受 GC 偏差的影响。
这些发现表明,在基因组测序中,由于 GC 偏差可能会导致潜在的困难,如果了解相关工作流程中固有的 GC 偏差,则可以通过方法优化来预先解决这些问题。此外,建议在宏基因组研究中对定量丰度估计采取更具批判性的方法。在未来,宏基因组研究应该在得出结论之前采取措施来考虑 GC 偏差的影响,或者使用证明无偏差的工作流程。