Folkhälsan Institute of Genetics, Folkhälsan Research Center, Biomedicum, Helsinki, Finland.
Department of Medical and Clinical Genetics, Medicum, University of Helsinki, Helsinki, Finland.
Acta Neuropathol Commun. 2020 Feb 17;8(1):18. doi: 10.1186/s40478-020-0893-1.
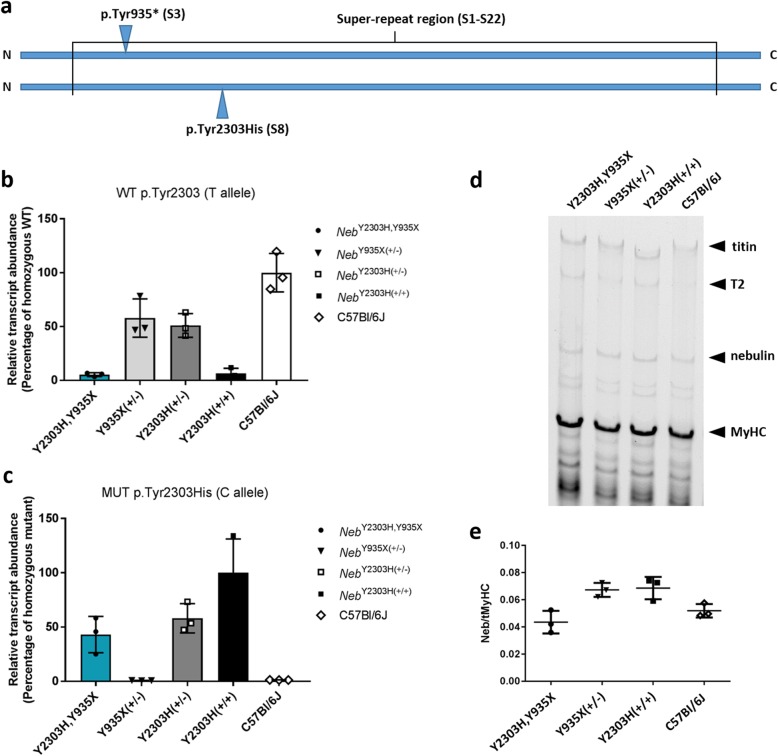
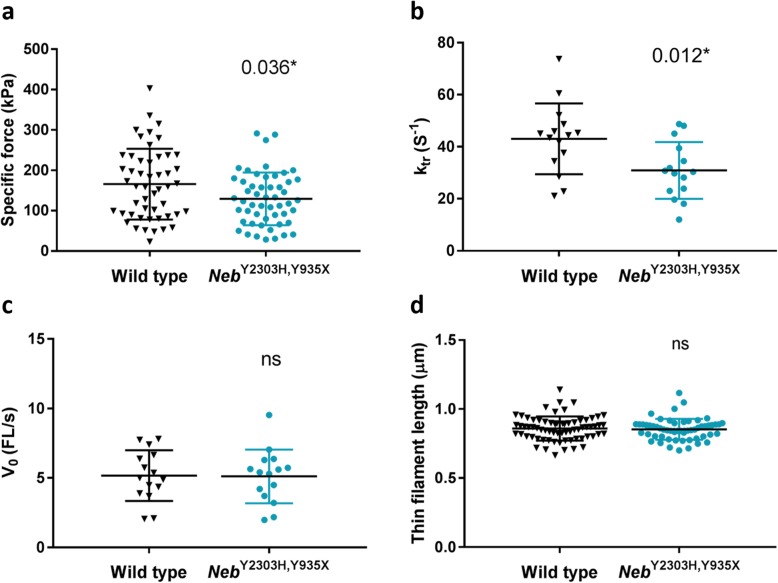
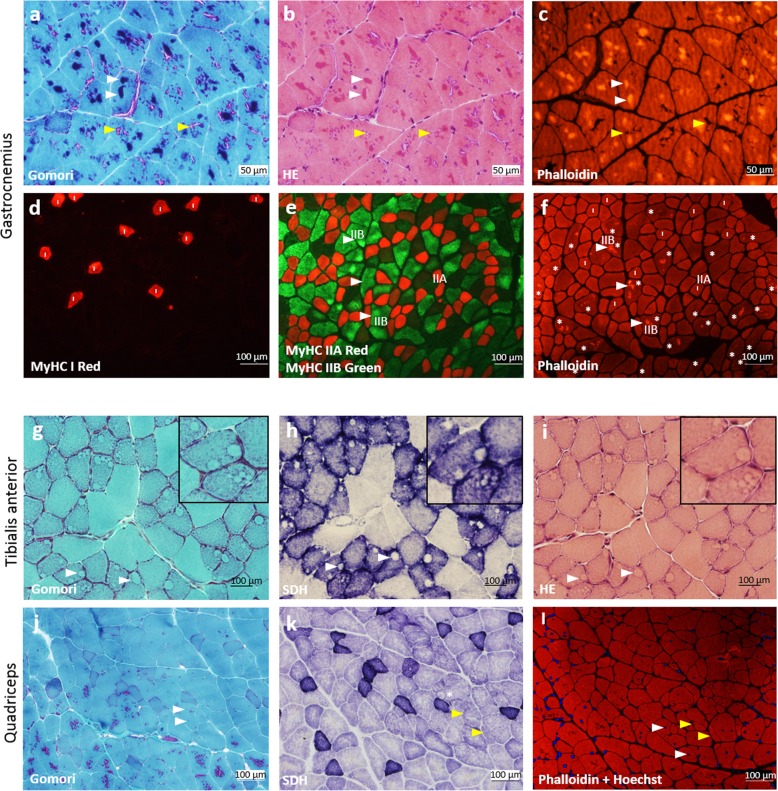
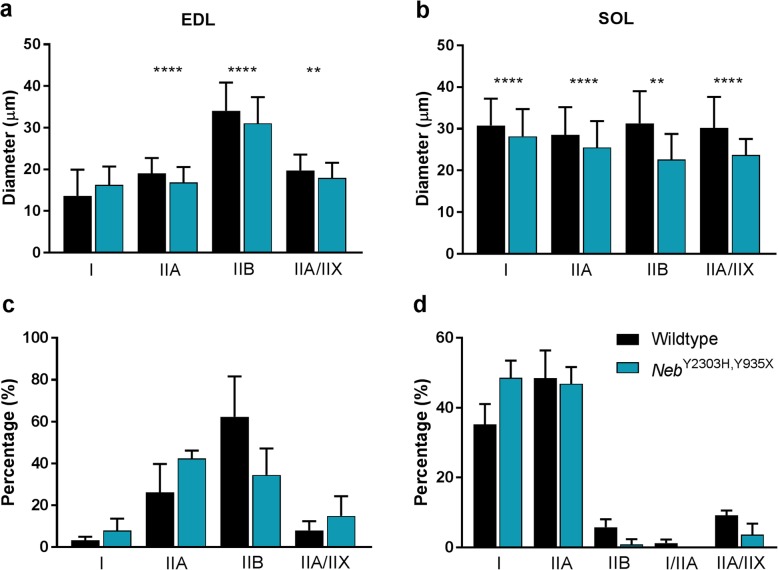
Nemaline myopathy (NM) caused by mutations in the gene encoding nebulin (NEB) accounts for at least 50% of all NM cases worldwide, representing a significant disease burden. Most NEB-NM patients have autosomal recessive disease due to a compound heterozygous genotype. Of the few murine models developed for NEB-NM, most are Neb knockout models rather than harbouring Neb mutations. Additionally, some models have a very severe phenotype that limits their application for evaluating disease progression and potential therapies. No existing murine models possess compound heterozygous Neb mutations that reflect the genotype and resulting phenotype present in most patients. We aimed to develop a murine model that more closely matched the underlying genetics of NEB-NM, which could assist elucidation of the pathogenetic mechanisms underlying the disease. Here, we have characterised a mouse strain with compound heterozygous Neb mutations; one missense (p.Tyr2303His), affecting a conserved actin-binding site and one nonsense mutation (p.Tyr935*), introducing a premature stop codon early in the protein. Our studies reveal that this compound heterozygous model, Neb, has striking skeletal muscle pathology including nemaline bodies. In vitro whole muscle and single myofibre physiology studies also demonstrate functional perturbations. However, no reduction in lifespan was noted. Therefore, Neb mice recapitulate human NEB-NM and are a much needed addition to the NEB-NM mouse model collection. The moderate phenotype also makes this an appropriate model for studying NEB-NM pathogenesis, and could potentially be suitable for testing therapeutic applications.
肌球蛋白结合蛋白 C 型 nemaline 肌病(NEB-NM)由 nebulin 基因(NEB)编码突变引起,占全球所有 NM 病例的至少 50%,代表着巨大的疾病负担。大多数 NEB-NM 患者由于复合杂合基因型而患有常染色体隐性疾病。在为 NEB-NM 开发的少数几种鼠模型中,大多数是 Neb 敲除模型,而不是具有 Neb 突变。此外,一些模型具有非常严重的表型,限制了它们在评估疾病进展和潜在疗法中的应用。没有现有的具有反映大多数患者基因型和表型的复合杂合 Neb 突变的鼠模型。我们旨在开发一种与 NEB-NM 潜在遗传学更匹配的鼠模型,这有助于阐明疾病的发病机制。在这里,我们描述了一种具有复合杂合 Neb 突变的小鼠品系;一个错义突变(p.Tyr2303His),影响保守的肌动蛋白结合位点,一个无义突变(p.Tyr935*),在蛋白的早期引入一个提前终止密码子。我们的研究表明,这种复合杂合模型 Neb 具有明显的骨骼肌病理学,包括 nemaline 体。体外整块肌肉和单个肌纤维生理学研究也表明存在功能障碍。然而,未观察到寿命缩短。因此,Neb 小鼠再现了人类的 NEB-NM,是 NEB-NM 鼠模型库中非常需要的补充。中度表型也使它成为研究 NEB-NM 发病机制的合适模型,并且可能适合测试治疗应用。