Kidney Health Service and Conjoint Renal Research Laboratory, Royal Brisbane and Women's Hospital, Brisbane, Australia.
Institute for Molecular Bioscience, The University of Queensland, Brisbane, Australia.
BMC Nephrol. 2020 Feb 22;21(1):58. doi: 10.1186/s12882-020-01717-9.
Fabry disease (FD) is a rare, lysosomal storage disorder caused by the absence or deficiency of the enzyme alpha-galactosidase A (α-Gal A) that leads to the abnormal accumulation of the lipid globotriaosylceramide (GB3) in a variety of cell types and tissues throughout the body. FD has an x-linked inheritance pattern. Previously thought to be only carriers, females can also experience FD symptomatology. Symptoms vary in type and severity from patient to patient and tend to increase in severity with age. FD symptoms are non-specific and may be shared with those of other diseases. Misdiagnoses and diagnostic delays are common, often resulting in progressive, irreversible tissue damage. The estimated prevalence of FD in the general population is 1:40,000 to 1:117,000 individuals. However, it is estimated that the prevalence of FD in the dialysis population is 0.12 to 0.7%. Little is known about the prevalence of FD in the broader Chronic Kidney Disease (CKD) population.
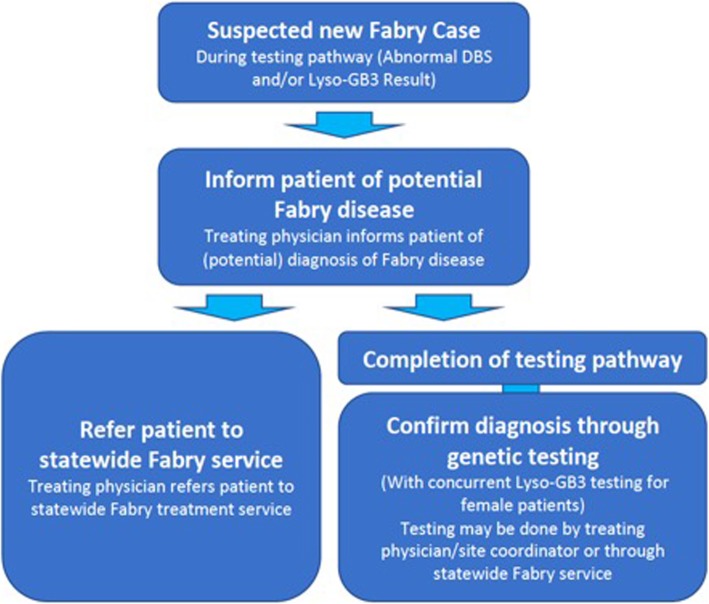
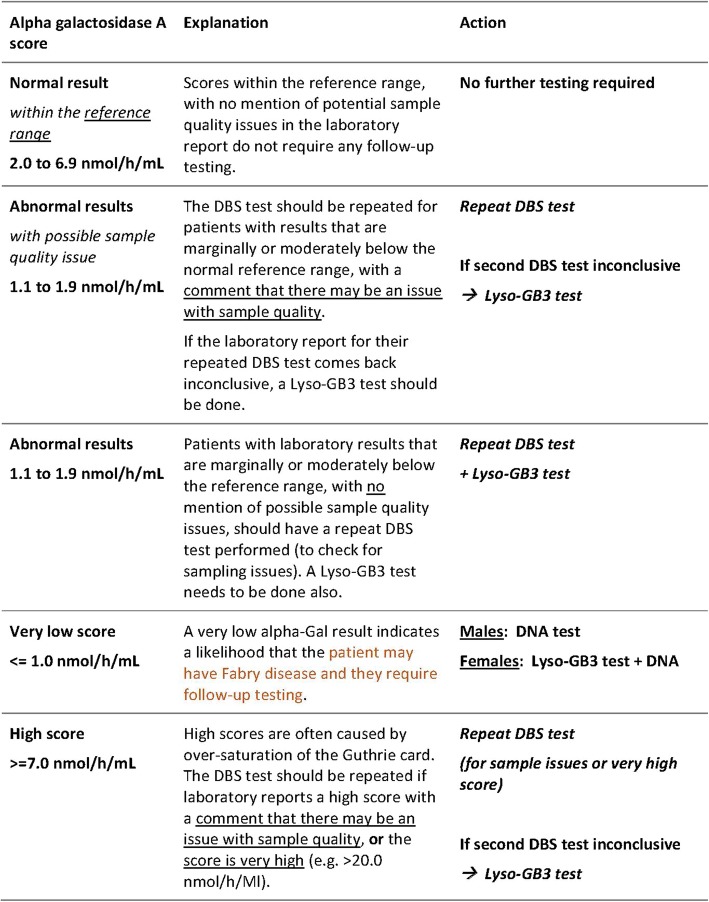
This is an epidemiological study of the prevalence of FD in CKD patents identified from the public renal speciality practices in Queensland, Australia. A cascade approach to screening is being employed with dried blood spot testing for blood levels of alpha-galactosidase A (Alpha-Gal), with follow-up testing for patients with abnormal results by plasma levels of globotriaosylsphingosine (Lyso-GB3) for females and non-definitive cases in males. A diagnosis of FD is confirmed through genetic testing of the GLA gene in cases suspected of having FD based upon Alpha-Gal and Lyso-GB3 testing.
Expected outcomes of this study include more information about the prevalence of FD at all stages of CKD, including for both males and females. The study may also provide information about common characteristics of FD to assist with diagnosis and optimal management/treatment. Screening is also available for family members of diagnosed patients, with potential for early diagnosis of FD and intervention for those individuals.
Queensland Health Database of Research Activity (DORA, https://dora.health.qld.gov.au) pj09946 (Registered 3rd July 2017).
法布里病(FD)是一种罕见的溶酶体贮积病,由α-半乳糖苷酶 A(α-Gal A)缺乏或缺失引起,导致脂质Globotriaosylceramide(GB3)在体内各种细胞类型和组织中异常积累。FD 具有 X 连锁遗传模式。以前认为只有女性是携带者,也可能出现 FD 症状。患者的症状类型和严重程度各不相同,且随着年龄的增长而趋于加重。FD 症状不具特异性,可能与其他疾病的症状相似。误诊和诊断延误很常见,常导致进行性、不可逆转的组织损伤。普通人群中 FD 的估计患病率为每 40,000 至 117,000 人中有 1 例。然而,据估计,在透析人群中 FD 的患病率为 0.12 至 0.7%。关于更广泛的慢性肾脏病(CKD)人群中 FD 的患病率知之甚少。
这是一项在澳大利亚昆士兰州公共肾脏专科实践中发现的 CKD 患者 FD 患病率的流行病学研究。采用级联方法进行筛查,通过干血斑检测α-半乳糖苷酶 A(Alpha-Gal)的血液水平,对结果异常的女性和男性非明确病例进行血浆 globotriaosylsphingosine(Lyso-GB3)检测。对疑似 FD 的患者,根据 Alpha-Gal 和 Lyso-GB3 检测结果,通过 GLA 基因的基因检测,对 FD 进行确诊。
本研究的预期结果包括有关 CKD 所有阶段 FD 患病率的更多信息,包括男性和女性。该研究还可能提供有关 FD 常见特征的信息,以协助诊断和优化管理/治疗。也可为确诊患者的家属提供筛查服务,为这些个体提供 FD 的早期诊断和干预机会。
昆士兰州卫生数据库的研究活动(DORA,https://dora.health.qld.gov.au)pj09946(2017 年 7 月 3 日注册)。