Information School, University of Sheffield, Regent Court, 211 Portobello, Sheffield, S1 4DP, UK.
Evotec (U.K.) Ltd, 114 Innovation Drive, Milton Park, Abingdon, OX14 4RZ, UK.
J Comput Aided Mol Des. 2020 Jul;34(7):783-803. doi: 10.1007/s10822-020-00300-6. Epub 2020 Feb 28.
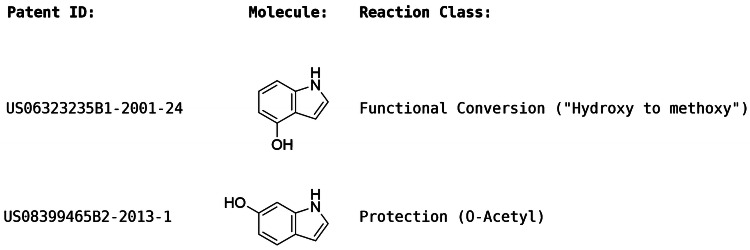
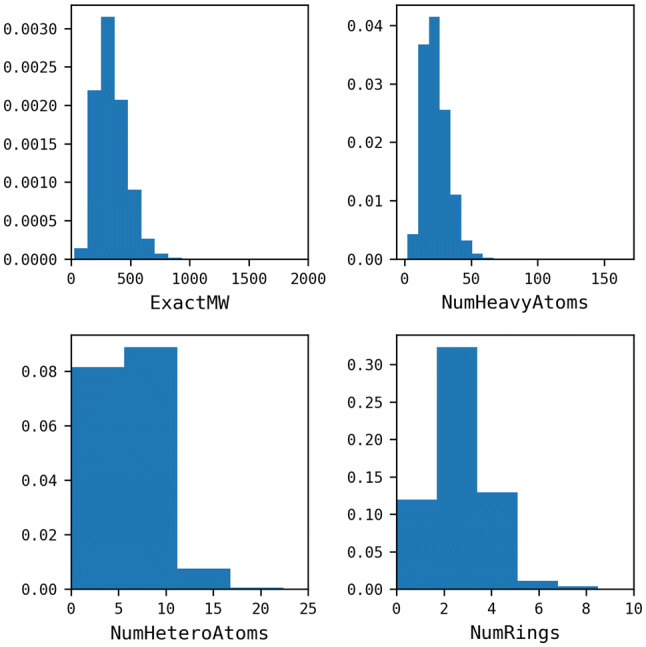
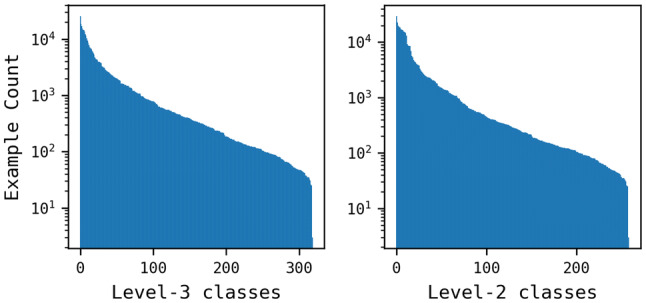
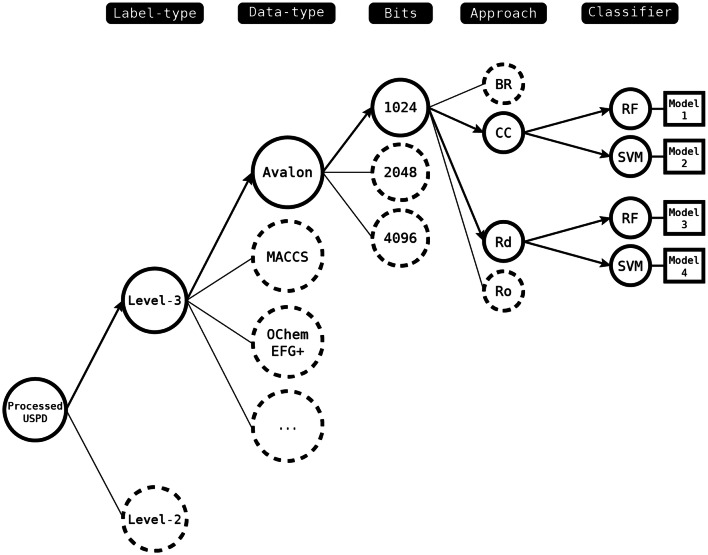
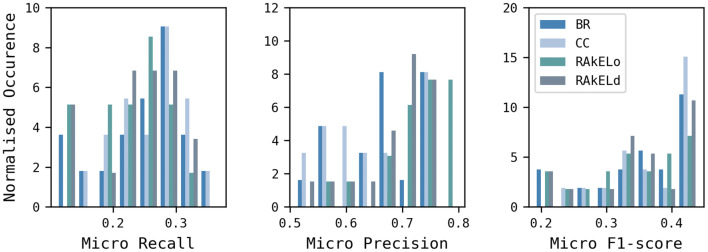
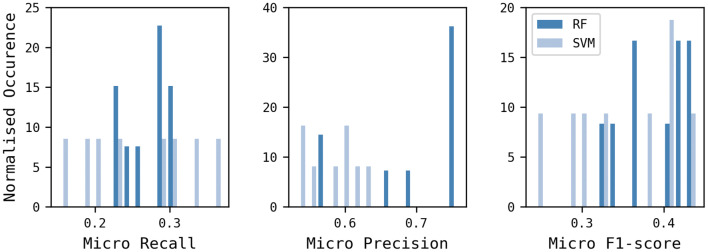
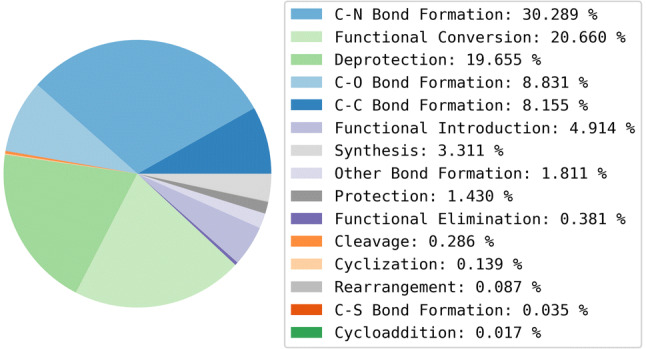
Reaction-based de novo design refers to the in-silico generation of novel chemical structures by combining reagents using structural transformations derived from known reactions. The driver for using reaction-based transformations is to increase the likelihood of the designed molecules being synthetically accessible. We have previously described a reaction-based de novo design method based on reaction vectors which are transformation rules that are encoded automatically from reaction databases. A limitation of reaction vectors is that they account for structural changes that occur at the core of a reaction only, and they do not consider the presence of competing functionalities that can compromise the reaction outcome. Here, we present the development of a Reaction Class Recommender to enhance the reaction vector framework. The recommender is intended to be used as a filter on the reaction vectors that are applied during de novo design to reduce the combinatorial explosion of in-silico molecules produced while limiting the generated structures to those which are most likely to be synthesisable. The recommender has been validated using an external data set extracted from the recent medicinal chemistry literature and in two simulated de novo design experiments. Results suggest that the use of the recommender drastically reduces the number of solutions explored by the algorithm while preserving the chance of finding relevant solutions and increasing the global synthetic accessibility of the designed molecules.
基于反应的从头设计是指通过使用结构转换来组合试剂,从而在计算机上生成新的化学结构,这些结构转换源自已知反应。使用基于反应的转换的驱动因素是增加所设计分子具有合成可及性的可能性。我们之前描述了一种基于反应向量的基于反应的从头设计方法,反应向量是从反应数据库自动编码的转换规则。反应向量的一个限制是,它们仅考虑反应核心发生的结构变化,而不考虑可能影响反应结果的竞争功能基团的存在。在这里,我们提出了一种反应类推荐器的开发,以增强反应向量框架。该推荐器旨在用作在从头设计中应用的反应向量的过滤器,以减少在计算机上生成的分子的组合爆炸,同时将生成的结构限制在最有可能可合成的结构。该推荐器已使用从最近的药物化学文献中提取的外部数据集和两个模拟从头设计实验进行了验证。结果表明,使用推荐器可以大大减少算法探索的解决方案数量,同时保留找到相关解决方案的机会,并增加所设计分子的全球合成可及性。