Venkatraman Vishwesh, Colligan Thomas H, Lesica George T, Olson Daniel R, Gaiser Jeremiah, Copeland Conner J, Wheeler Travis J, Roy Amitava
Department of Chemistry, Norwegian University of Science and Technology, Trondheim, Norway.
Department of Computer Science, University of Montana, Missoula, MT, United States.
Front Pharmacol. 2022 Apr 26;13:874746. doi: 10.3389/fphar.2022.874746. eCollection 2022.
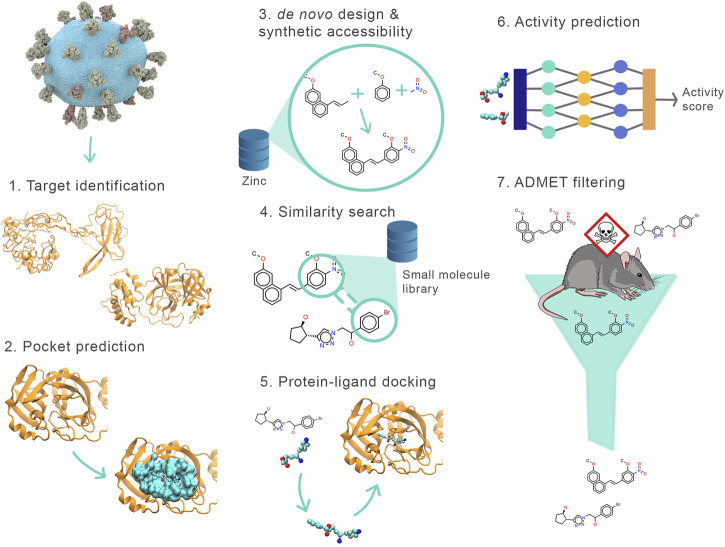
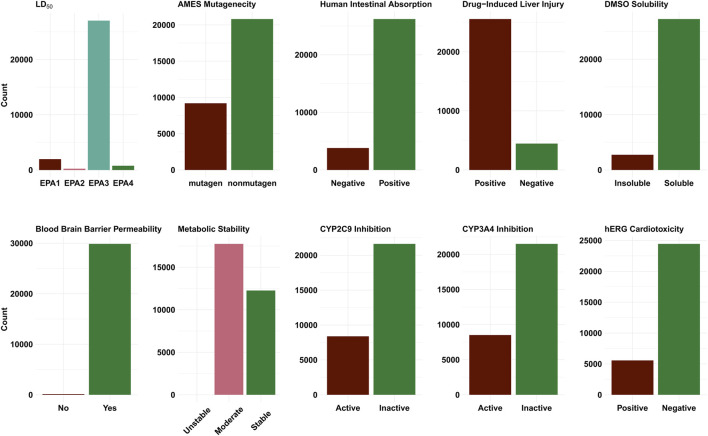
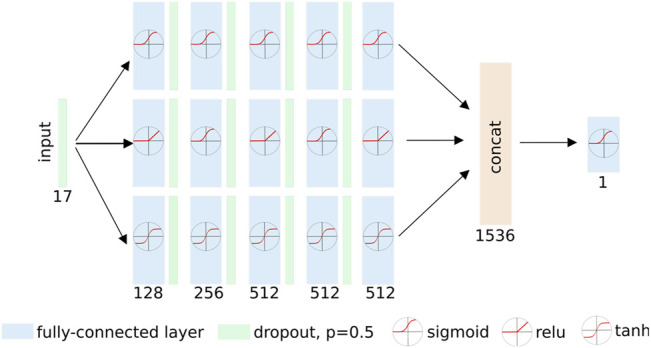
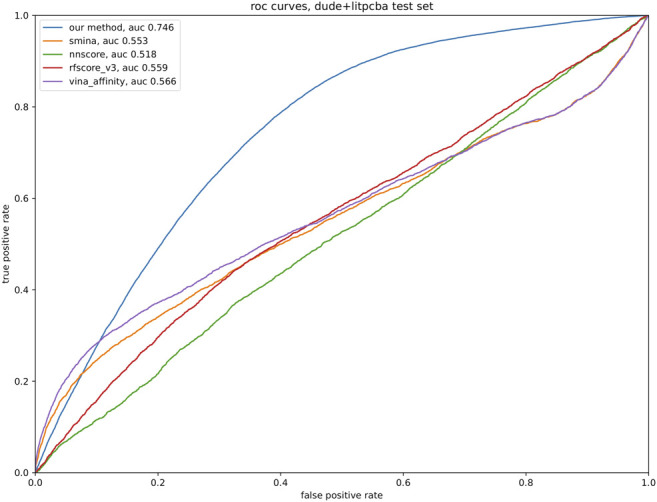
The SARS-CoV2 pandemic has highlighted the importance of efficient and effective methods for identification of therapeutic drugs, and in particular has laid bare the need for methods that allow exploration of the full diversity of synthesizable small molecules. While classical high-throughput screening methods may consider up to millions of molecules, virtual screening methods hold the promise of enabling appraisal of billions of candidate molecules, thus expanding the search space while concurrently reducing costs and speeding discovery. Here, we describe a new screening pipeline, called , that is capable of rapidly exploring drug candidates from a library of billions of molecules, and is designed to support distributed computation on cluster and cloud resources. As an example of performance, our pipeline required ∼40,000 total compute hours to screen for potential drugs targeting three SARS-CoV2 proteins among a library of ∼3.7 billion candidate molecules.
严重急性呼吸综合征冠状病毒2(SARS-CoV-2)大流行凸显了高效识别治疗药物方法的重要性,尤其暴露了对能够探索可合成小分子全部多样性的方法的需求。虽然传统的高通量筛选方法可能会考虑多达数百万种分子,但虚拟筛选方法有望评估数十亿种候选分子,从而扩大搜索空间,同时降低成本并加速发现进程。在此,我们描述了一种名为 的新筛选流程,它能够从数十亿分子的库中快速探索候选药物,并设计用于支持在集群和云资源上进行分布式计算。作为性能示例,我们的流程在约37亿个候选分子库中筛选针对三种SARS-CoV-2蛋白的潜在药物总共需要约40000个计算小时。