Hall Thomas J, Vernimmen Douglas, Browne John A, Mullen Michael P, Gordon Stephen V, MacHugh David E, O'Doherty Alan M
Animal Genomics Laboratory, UCD School of Agriculture and Food Science, College Dublin, Dublin, Ireland.
The Roslin Institute and Royal (Dick) School of Veterinary Studies, University of Edinburgh, Easter Bush, Midlothian, United Kingdom.
Front Genet. 2020 Feb 7;10:1386. doi: 10.3389/fgene.2019.01386. eCollection 2019.
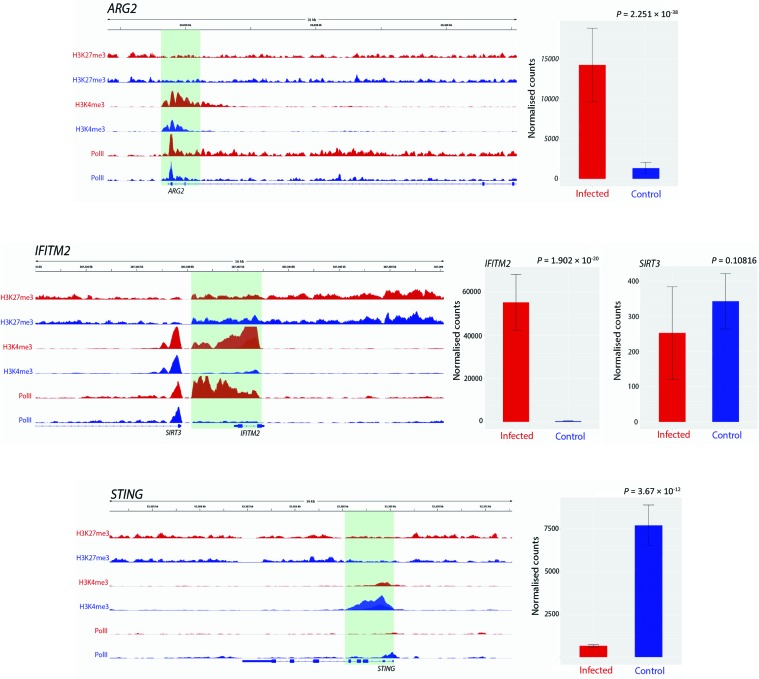
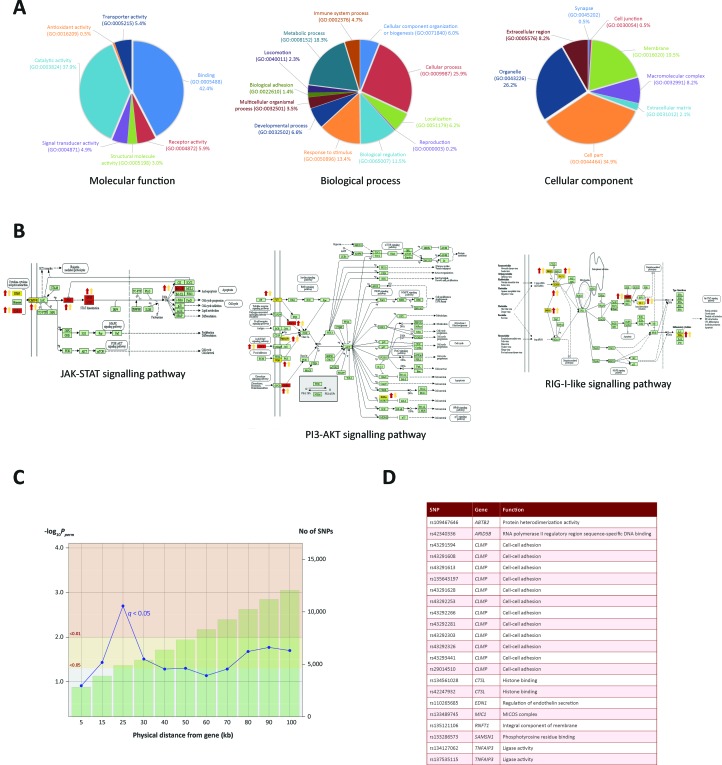
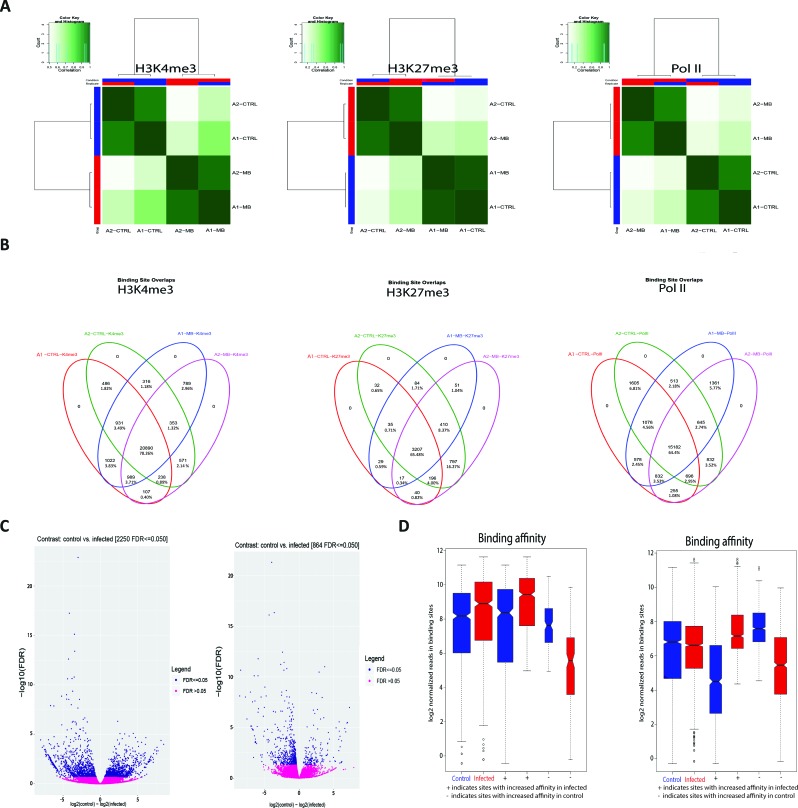
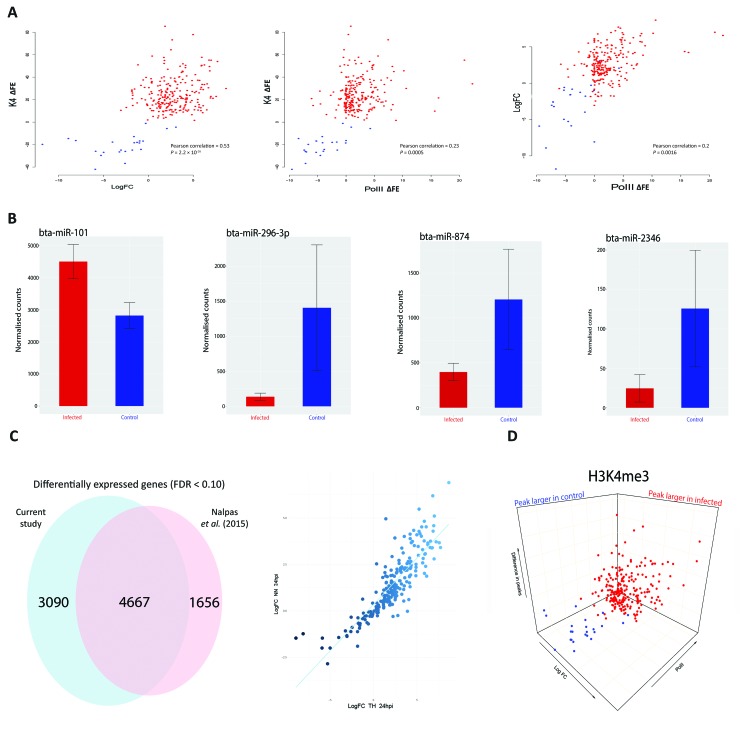
Bovine tuberculosis is caused by infection with , which can also cause disease in a range of other mammals, including humans. Alveolar macrophages are the key immune effector cells that first encounter and how the macrophage epigenome responds to mycobacterial pathogens is currently not well understood. Here, we have used chromatin immunoprecipitation sequencing (ChIP-seq), RNA-seq and miRNA-seq to examine the effect of infection on the bovine alveolar macrophage (bAM) epigenome. We show that H3K4me3 is more prevalent, at a genome-wide level, in chromatin from -infected bAM compared to control non-infected bAM; this was particularly evident at the transcriptional start sites of genes that determine programmed macrophage responses to mycobacterial infection (e.g. M1/M2 macrophage polarisation). This pattern was also supported by the distribution of RNA Polymerase II (Pol II) ChIP-seq results, which highlighted significantly increased transcriptional activity at genes demarcated by permissive chromatin. Identification of these genes enabled integration of high-density genome-wide association study (GWAS) data, which revealed genomic regions associated with resilience to infection with in cattle. Through integration of these data, we show that bAM transcriptional reprogramming occurs through differential distribution of H3K4me3 and Pol II at key immune genes. Furthermore, this subset of genes can be used to prioritise genomic variants from a relevant GWAS data set.
牛结核病是由感染 引起的, 也可在包括人类在内的一系列其他哺乳动物中引发疾病。肺泡巨噬细胞是首先接触 的关键免疫效应细胞,而巨噬细胞表观基因组如何响应分枝杆菌病原体目前尚不清楚。在这里,我们使用染色质免疫沉淀测序(ChIP-seq)、RNA测序(RNA-seq)和微小RNA测序(miRNA-seq)来研究 感染对牛肺泡巨噬细胞(bAM)表观基因组的影响。我们发现,在全基因组水平上,与未感染的对照bAM相比,感染 的bAM染色质中H3K4me3更为普遍;这在决定巨噬细胞对分枝杆菌感染的程序性反应的基因(如M1/M2巨噬细胞极化)的转录起始位点尤为明显。RNA聚合酶II(Pol II)的ChIP-seq结果分布也支持了这一模式,该结果突出了由宽松染色质划定的基因处转录活性显著增加。对这些基因的鉴定使得能够整合高密度全基因组关联研究(GWAS)数据,从而揭示与牛抗 感染能力相关的基因组区域。通过整合这些数据,我们表明bAM转录重编程是通过关键免疫基因处H3K4me3和Pol II的差异分布发生的。此外,这一子集基因可用于对相关GWAS数据集中的基因组变异进行优先级排序。