Zhang Kaimin, Fan Chun, Cai Dongpeng, Zhang Yi, Zuo Rui, Zhu Li, Cao Yue, Zhang Jian, Liu Chao, Chen Yang, Liang Hui
School of Pharmaceutical, Guangzhou University of Chinese Medicine, Guangzhou, China.
Guangdong Provincial Hospital of Chinese Medicine, Guangzhou University of Chinese Medicine, Guangzhou, China.
Front Cell Dev Biol. 2020 Feb 5;8:1. doi: 10.3389/fcell.2020.00001. eCollection 2020.
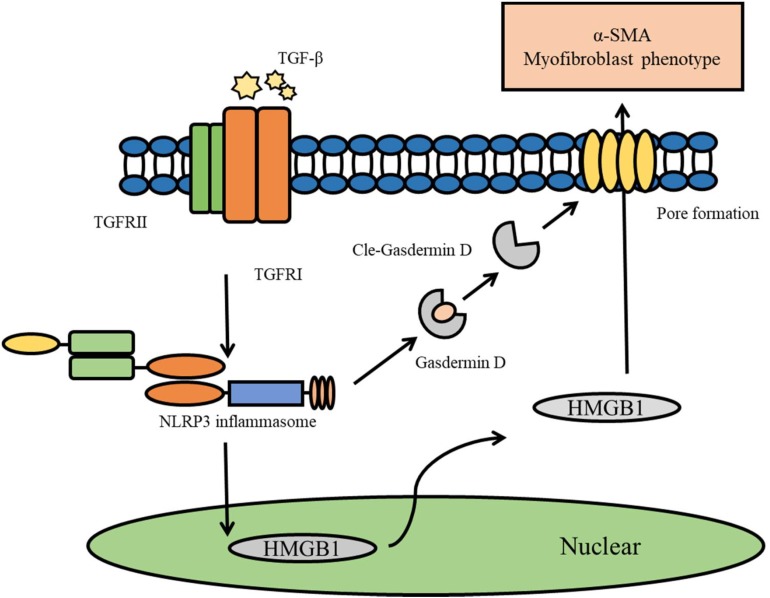
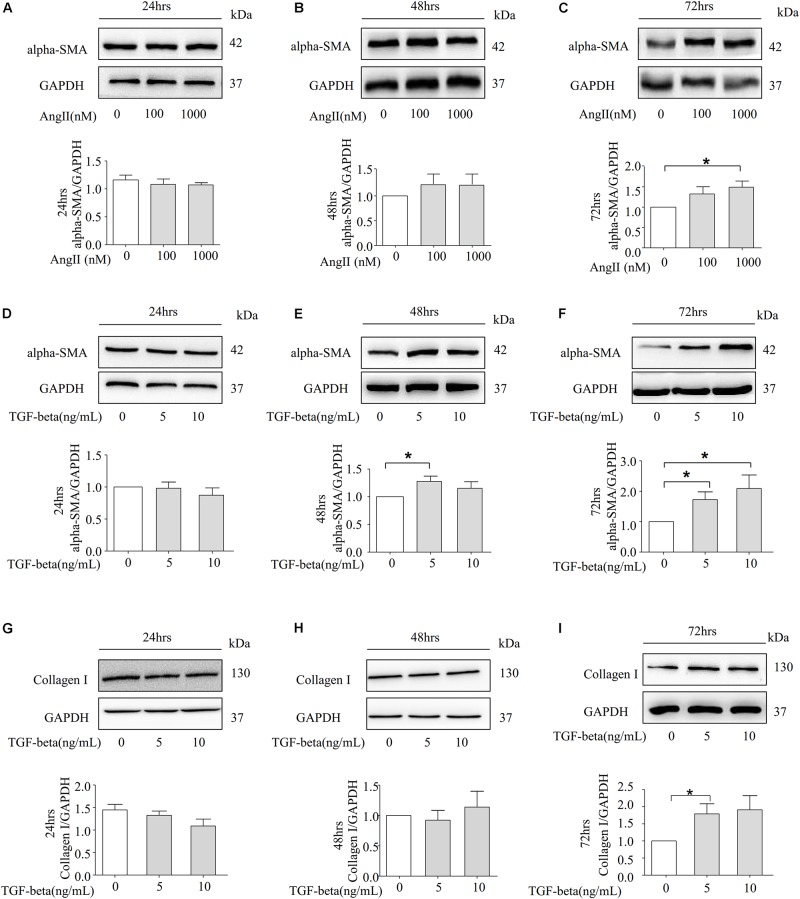
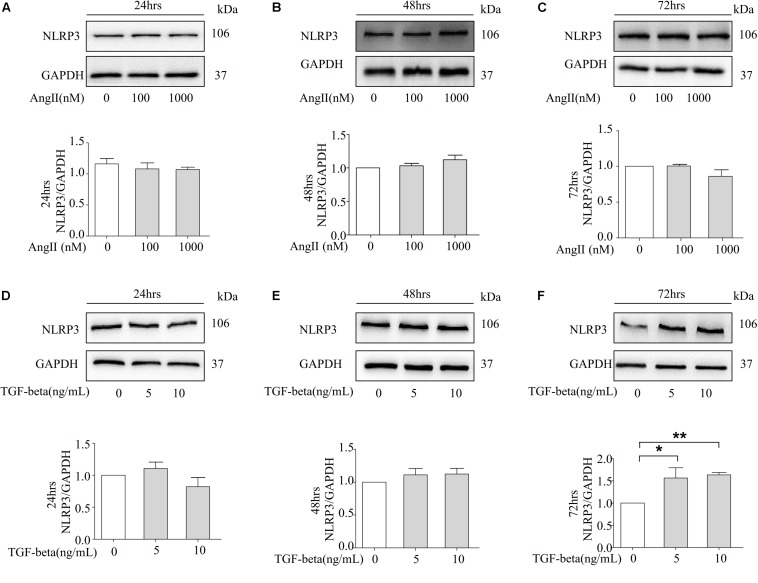
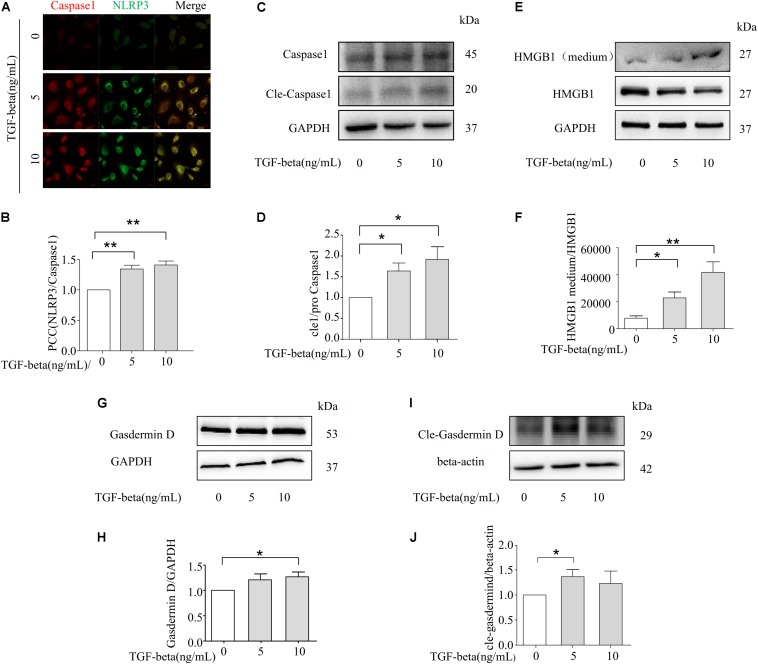
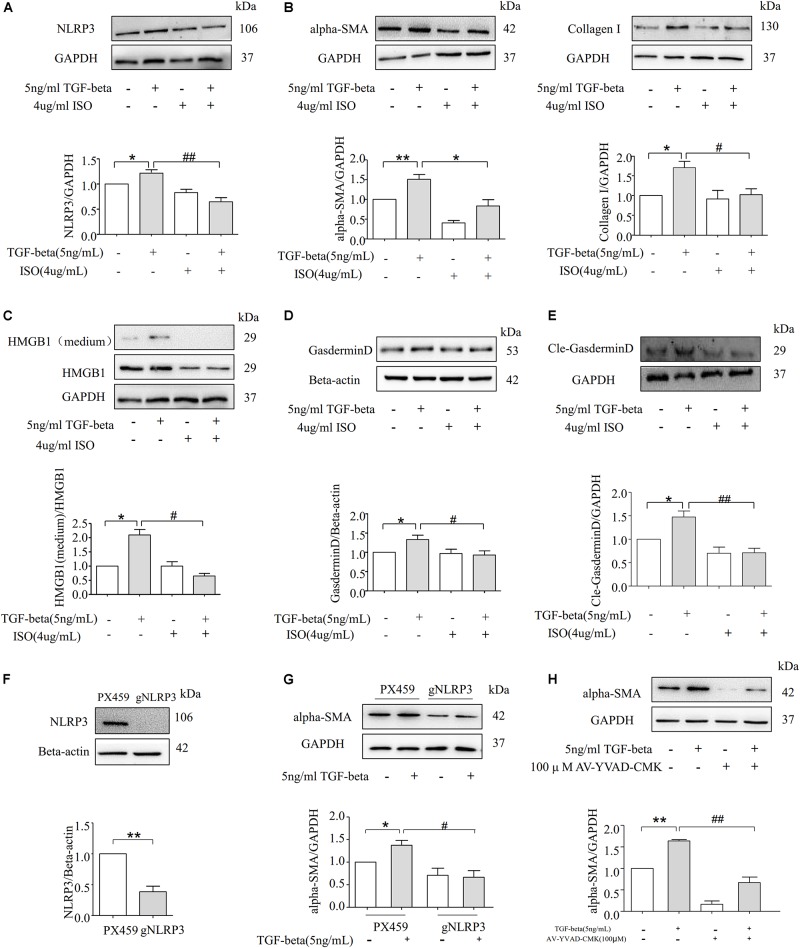
Fibrosis is a common phenotype that often leads to the progression of blood pressure-induced chronic kidney disease (CKD). TGF-beta plays an important role in promoting pathogenesis, and NLRP3 is a critical mediator in the progression of blood pressure-induced CKD. However, the pathophysiological roles of the TGF-beta-mediated NLRP3 pathway in modulating fibrosis in blood pressure-induced CKD have not been elucidated. The present study aims to investigate the contribution of TGF-beta-mediated NLRP3 inflammasome to renal fibrosis in rats with high blood pressure. By treating rats with angiotensin II (Ang II) for 14 days, we observed the development of fibrosis, characterized by epithelial-mesenchymal transition (EMT) markers [alpha-smooth muscle actin (alpha-SMA), MMP-2, and MMP-9]. Immunohistochemical analysis further revealed that TGF-beta and NLRP3 inflammasome activation [high-mobility group box 1 (HMGB1), IL-1beta, and NLRP3] were significantly upregulated in the kidney of rats with Ang II-induced hypertension. Interestingly, we observed that Ang II could not increase the production of NLRP3 proteins, but TGF-beta could induce NLRP3 protein expression in cultured NRK-52E cells. Furthermore, we speculated that TGF-beta played a pathogenic role in Ang II-induced CKD because TGF-beta induced the activation of NLRP3 inflammasomes and Gasdermin D cleavage expression. We also proved that the pharmacological inhibition of NLRP3 by ISO caused a decrease in TGF-beta-induced NLRP3 inflammasome activation and the expression of EMT markers (alpha-SMA and CollagenI) and Gasdermin D cleavage. Collectively, these results suggest that TGF-beta-mediated NLRP3 inflammasome activation may cause the release of HMGB1 and an increase in Gasdermin D cleavage in NRK-52E, thereby contributing to renal fibrosis in Ang II-induced CKD. These findings provide novel insights into the pathogenic role of NLRP3 in CKD associated with high blood pressure.
纤维化是一种常见的表型,常导致血压诱导的慢性肾脏病(CKD)进展。转化生长因子-β(TGF-β)在促进发病机制中起重要作用,而NLRP3是血压诱导的CKD进展中的关键介质。然而,TGF-β介导的NLRP3通路在调节血压诱导的CKD纤维化中的病理生理作用尚未阐明。本研究旨在探讨TGF-β介导的NLRP3炎性小体对高血压大鼠肾纤维化的作用。通过用血管紧张素II(Ang II)处理大鼠14天,我们观察到以上皮-间质转化(EMT)标志物[α-平滑肌肌动蛋白(α-SMA)、基质金属蛋白酶-2(MMP-2)和基质金属蛋白酶-9(MMP-9)]为特征的纤维化发展。免疫组织化学分析进一步显示,在Ang II诱导的高血压大鼠肾脏中,TGF-β和NLRP3炎性小体激活[高迁移率族蛋白B1(HMGB1)、白细胞介素-1β(IL-1β)和NLRP3]显著上调。有趣的是,我们观察到Ang II不能增加NLRP3蛋白的产生,但TGF-β可以在培养的NRK-52E细胞中诱导NLRP3蛋白表达。此外,我们推测TGF-β在Ang II诱导的CKD中起致病作用,因为TGF-β诱导NLRP3炎性小体激活和Gasdermin D裂解表达。我们还证明,ISO对NLRP3的药理抑制导致TGF-β诱导的NLRP3炎性小体激活以及EMT标志物(α-SMA和I型胶原蛋白)和Gasdermin D裂解表达的减少。总体而言,这些结果表明,TGF-β介导的NLRP3炎性小体激活可能导致HMGB1释放和NRK-52E中Gasdermin D裂解增加,从而导致Ang II诱导的CKD中的肾纤维化。这些发现为NLRP3在高血压相关CKD中的致病作用提供了新的见解。