Department of Urology, School of Medicine, The First Affiliated Hospital, Zhejiang University, Hangzhou, China.
J Cell Mol Med. 2020 Apr;24(7):4092-4104. doi: 10.1111/jcmm.15063. Epub 2020 Mar 3.
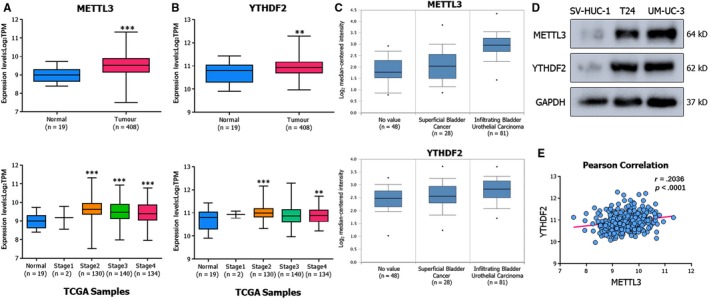
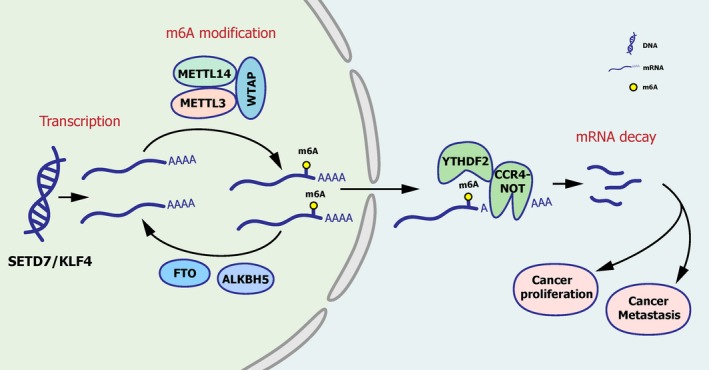
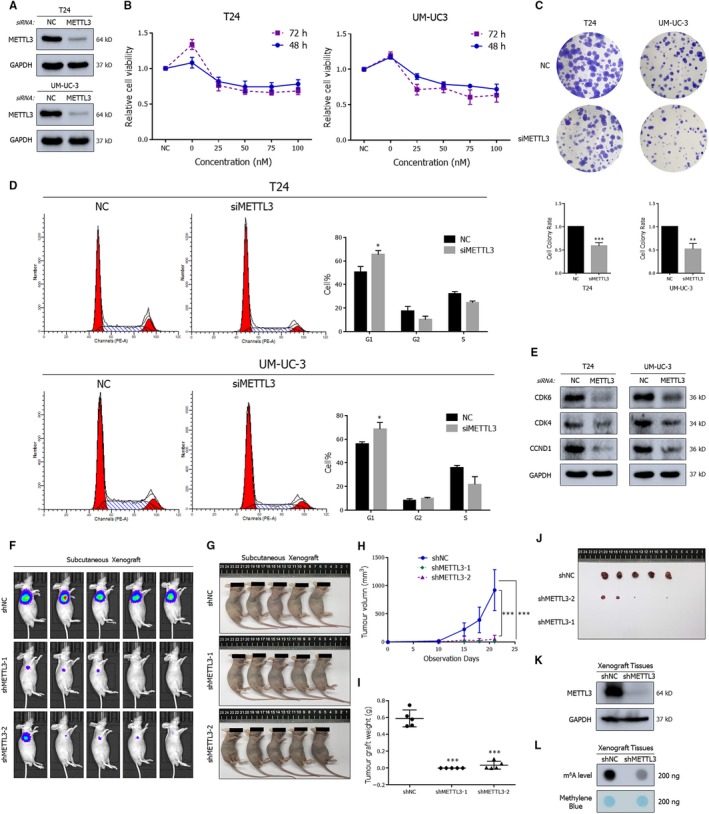
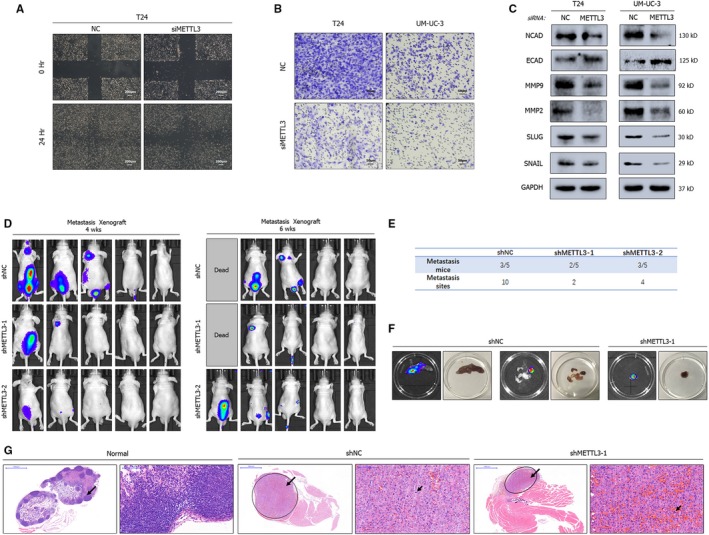
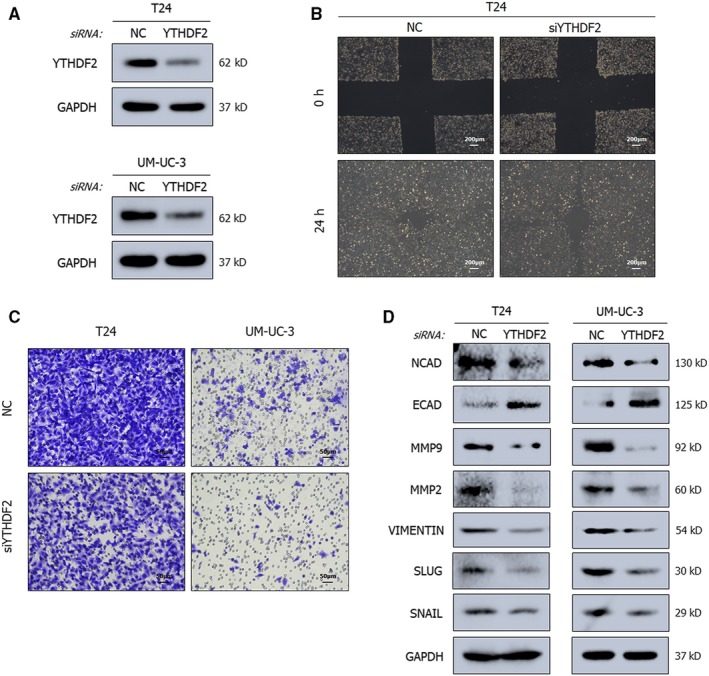
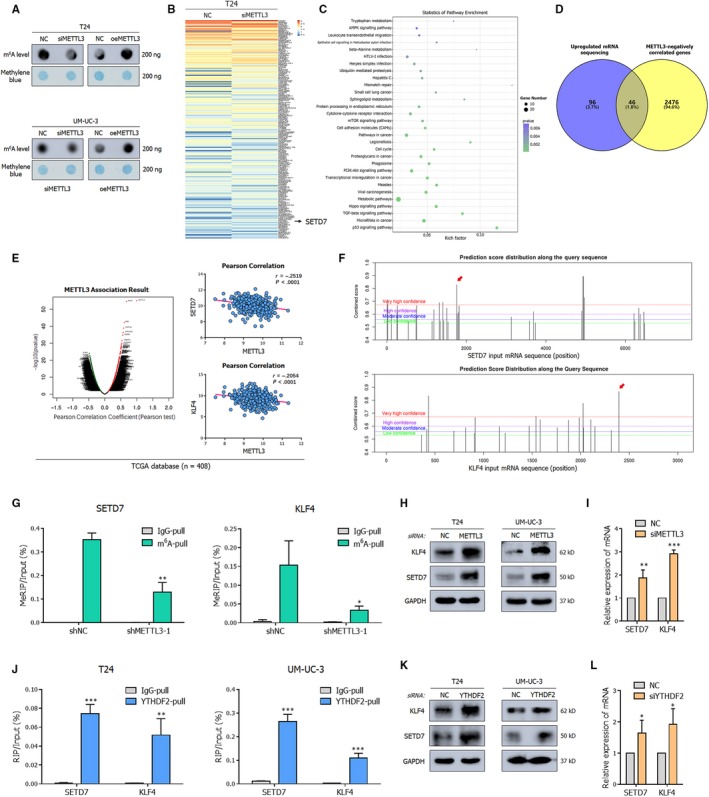
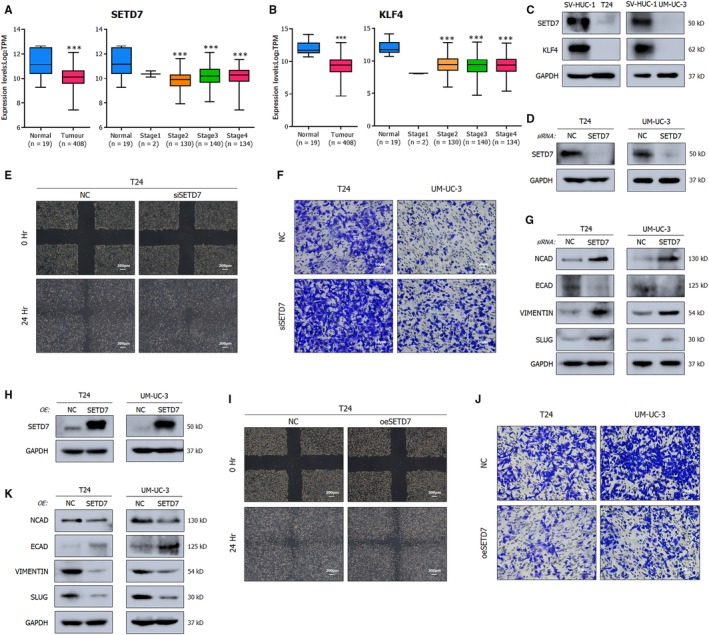
N6-Methyladenosine (m A) modification, the most prevalent modification of eukaryotic messenger RNA (mRNA), is involved in the progression of various tumours. However, the specific role of m A in bladder cancer (BCa) is still poorly understood. In this study, we demonstrated the tumour-promoting function and specific regulatory mechanism of m A axis, consisting of the core 'writer' protein METTL3 and the major reader protein YTHDF2. Depletion of METTL3 impaired cancer proliferation and cancer metastasis in vitro and in vivo. Through transcriptome sequencing, m A methylated RNA immunoprecipitation (MeRIP) and RIP, we determined that the METTL3/YTHDF2 m A axis directly degraded the mRNAs of the tumour suppressors SETD7 and KLF4, contributing to the progression of BCa. In addition, overexpression of SETD7 and KLF4 revealed a phenotype consistent with that induced by depletion of the m A axis. Thus, our findings on the METTL3/YTHDF2/SETD7/KLF4 m A axis provide the insight into the underlying mechanism of carcinogenesis and highlight potential therapeutic targets for BCa.
N6-甲基腺苷(m6A)修饰是真核信使 RNA(mRNA)中最普遍的修饰,参与多种肿瘤的进展。然而,m6A 在膀胱癌(BCa)中的具体作用仍知之甚少。在这项研究中,我们证明了由核心“书写器”蛋白 METTL3 和主要读码蛋白 YTHDF2 组成的 m6A 轴具有促进肿瘤的功能和特定的调节机制。METTL3 的耗竭会损害体外和体内的癌症增殖和癌症转移。通过转录组测序、m6A 甲基化 RNA 免疫沉淀(MeRIP)和 RIP,我们确定了 METTL3/YTHDF2 m6A 轴直接降解肿瘤抑制因子 SETD7 和 KLF4 的 mRNA,促进了 BCa 的进展。此外,SETD7 和 KLF4 的过表达显示出与 m6A 轴耗竭诱导的表型一致。因此,我们关于 METTL3/YTHDF2/SETD7/KLF4 m6A 轴的发现提供了对致癌发生潜在机制的深入了解,并强调了 BCa 的潜在治疗靶点。