Department of Oncology, Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China.
Department of Obstetrics and Gynecology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei 430030, China.
Biomed Res Int. 2020 Feb 28;2020:4169691. doi: 10.1155/2020/4169691. eCollection 2020.
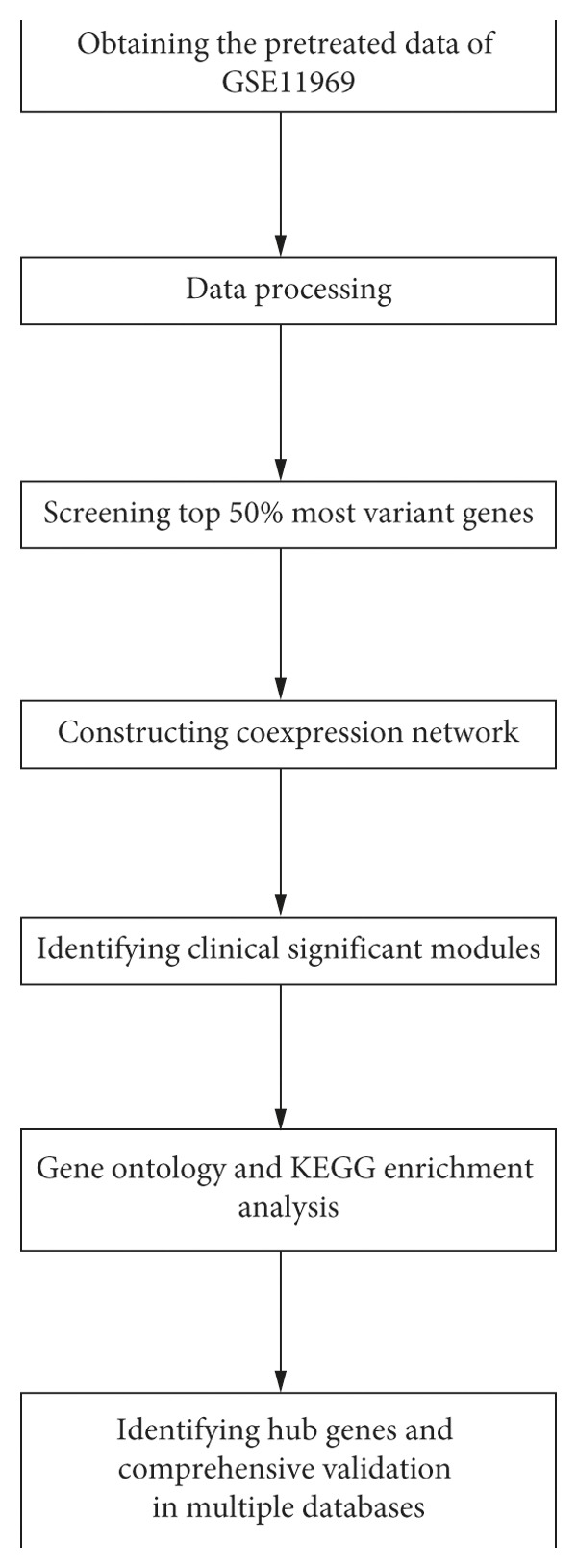
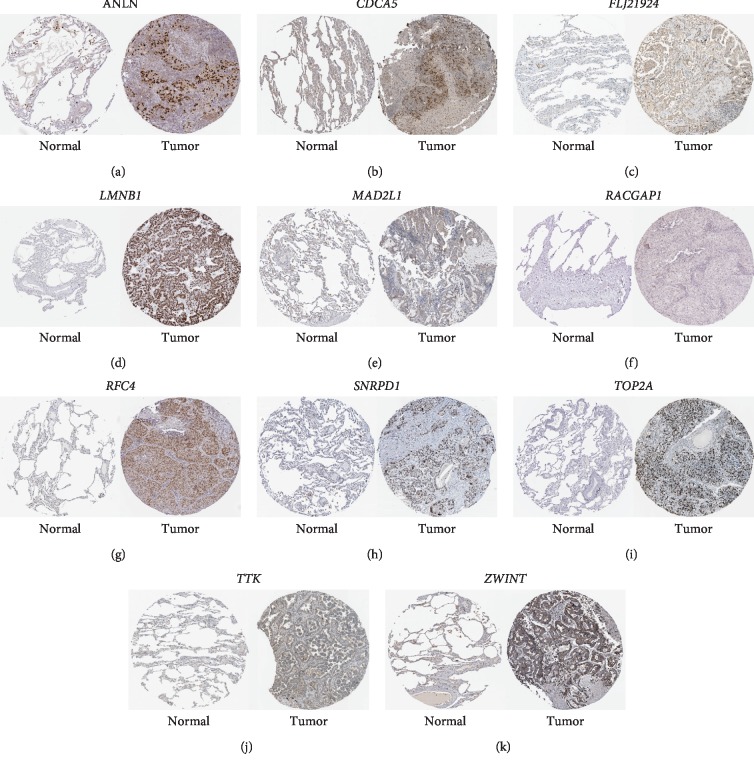
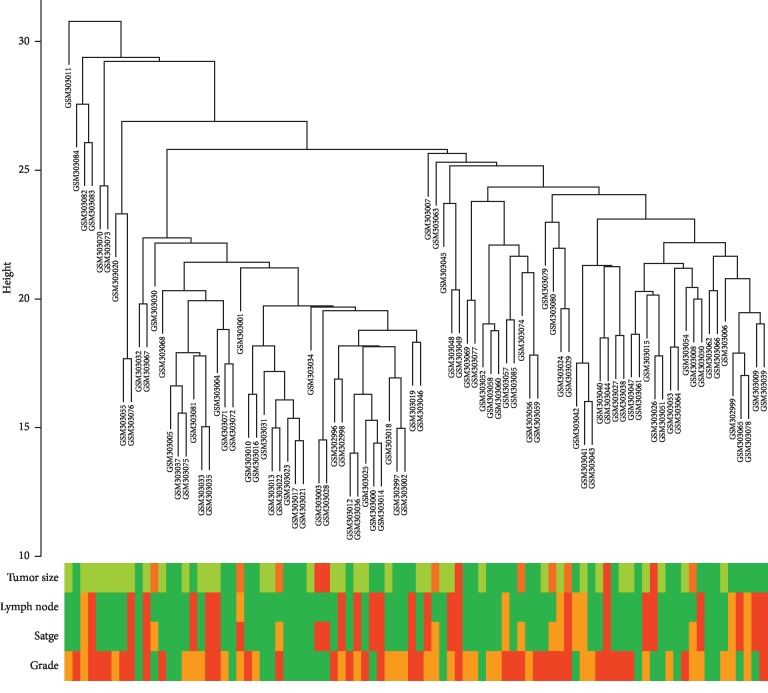
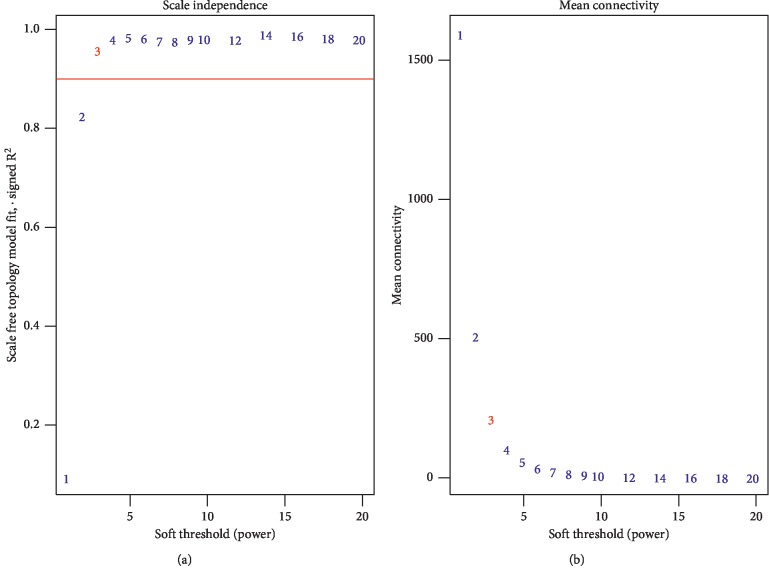
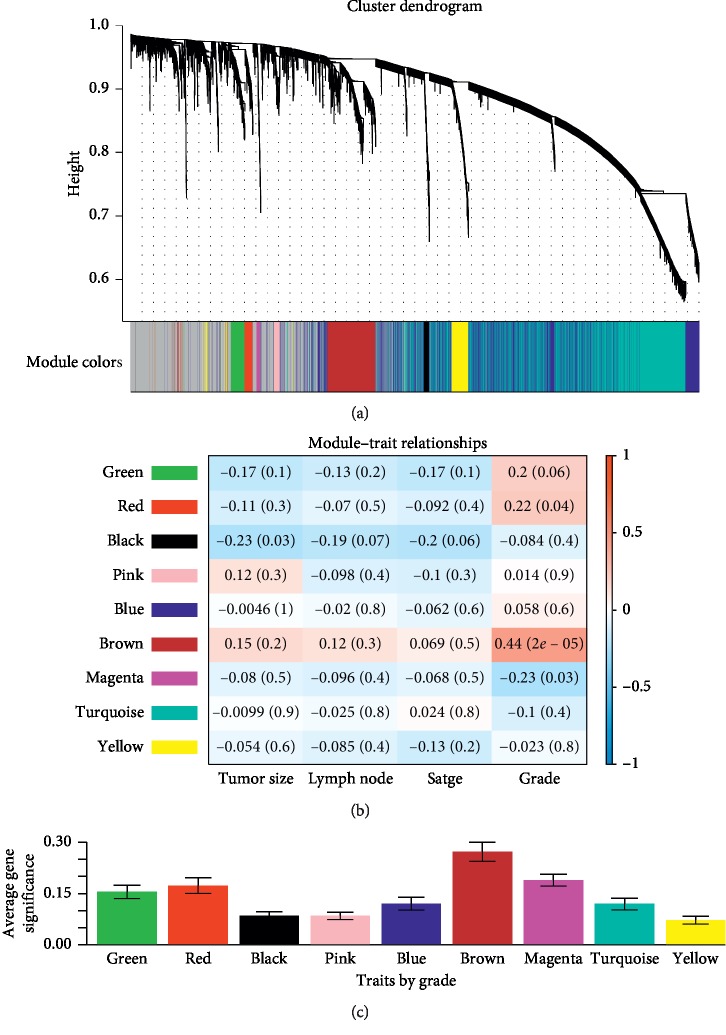
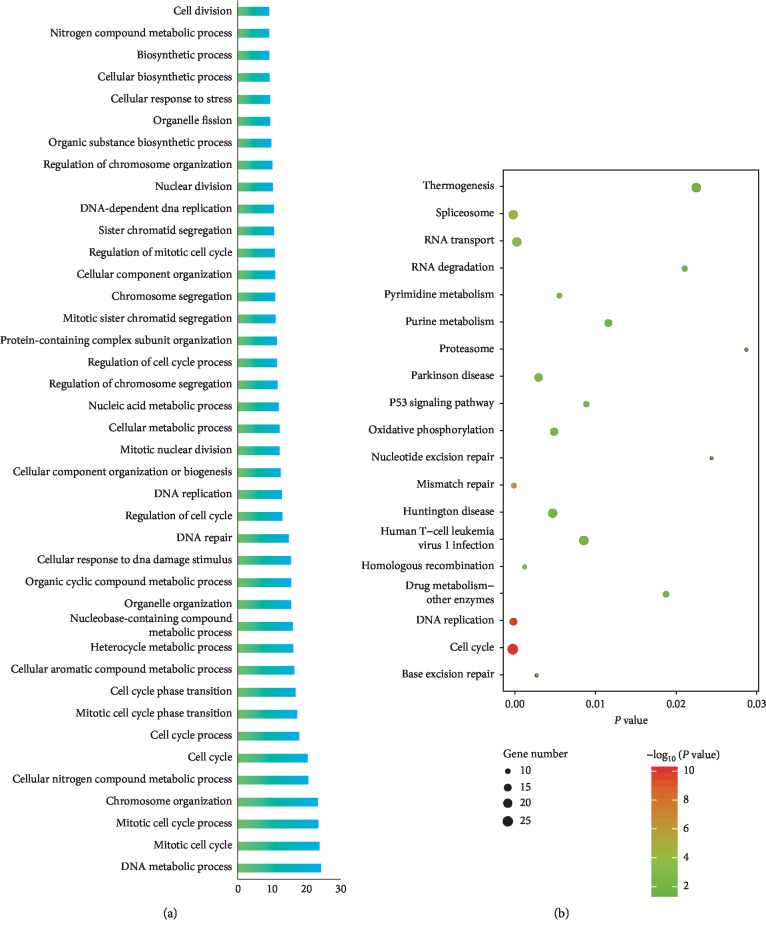
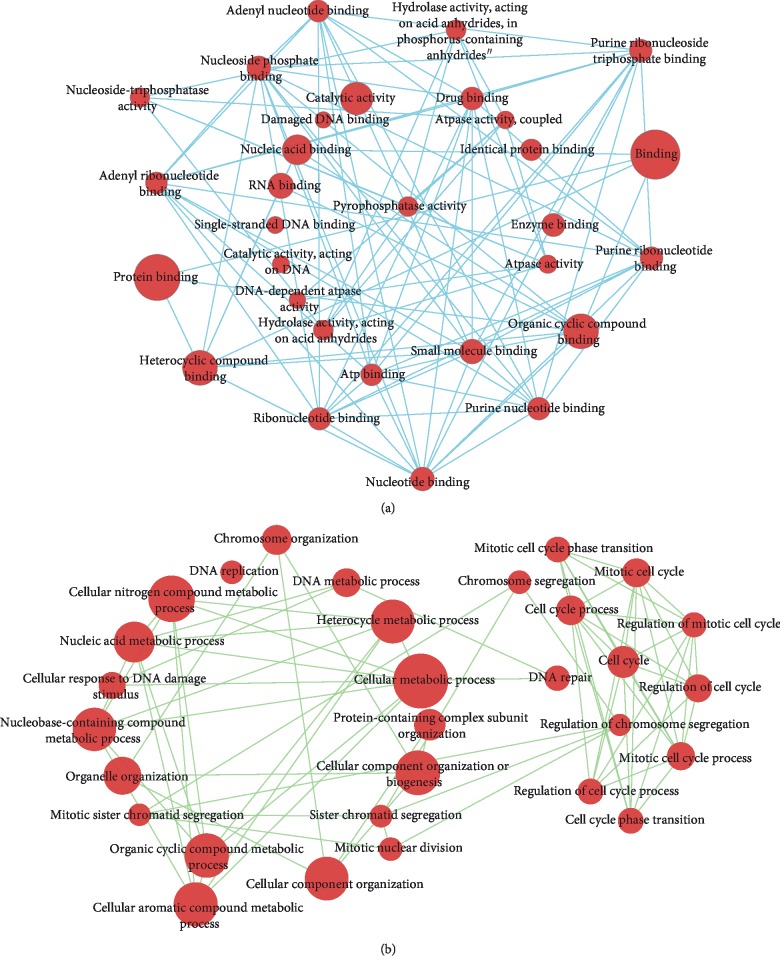
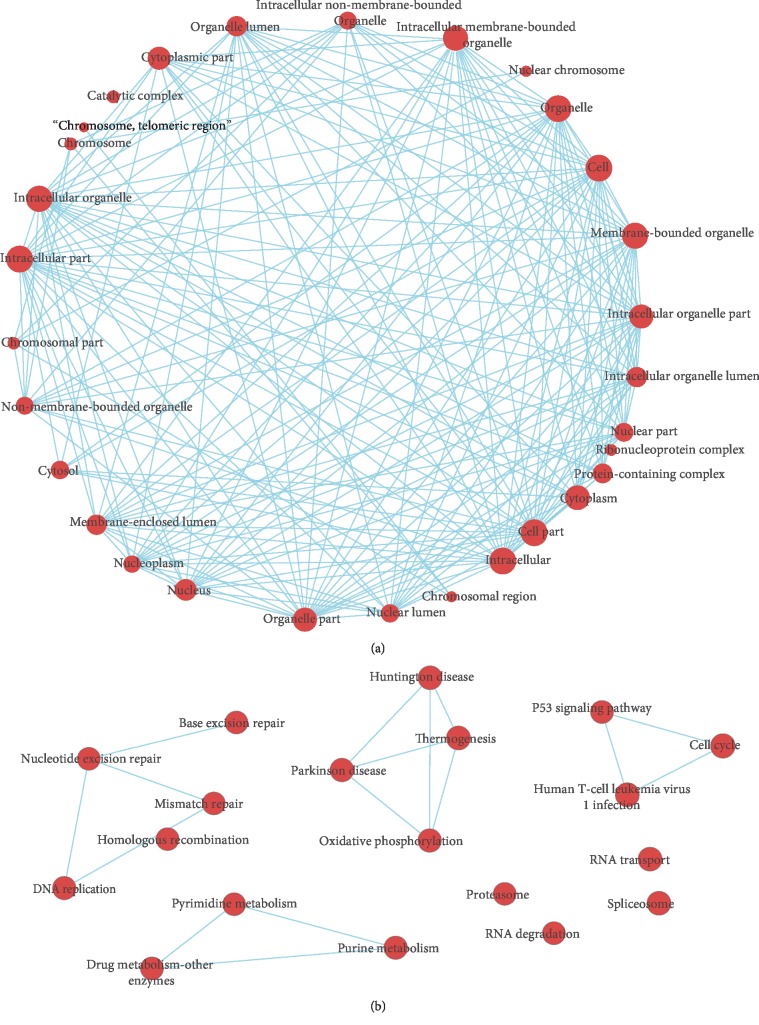
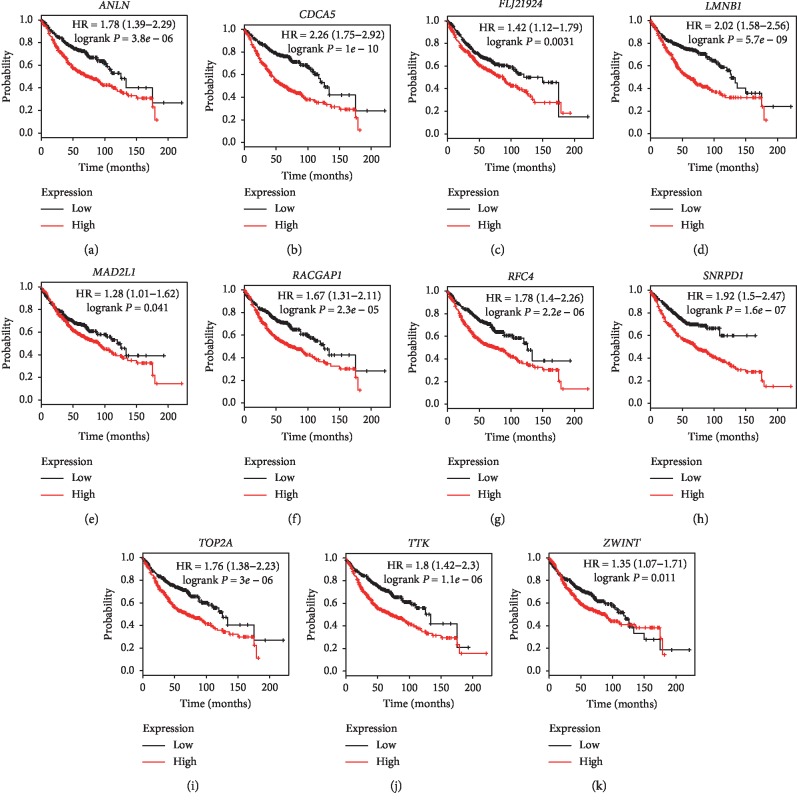
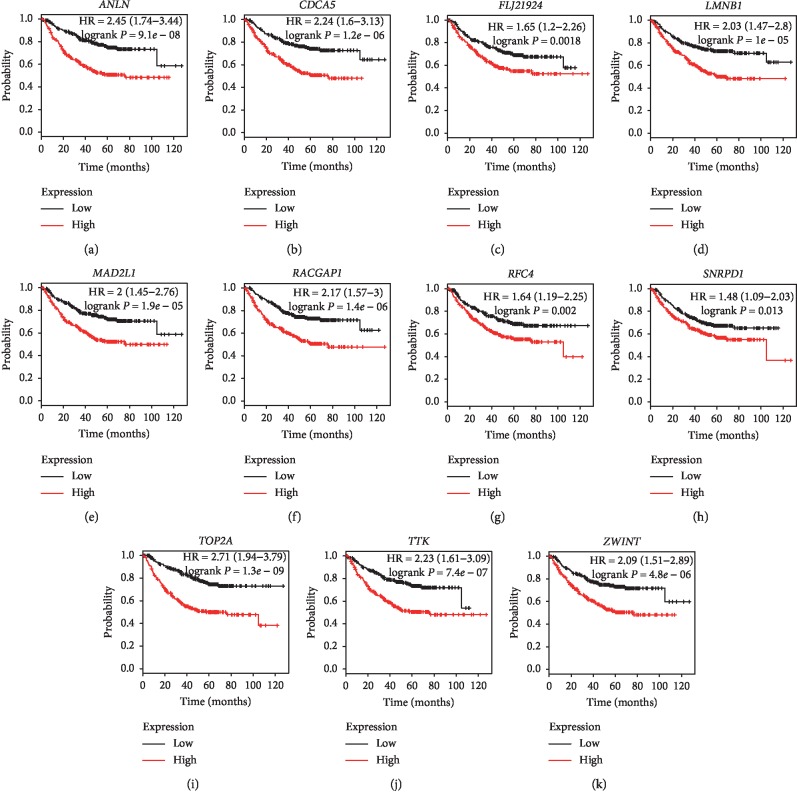
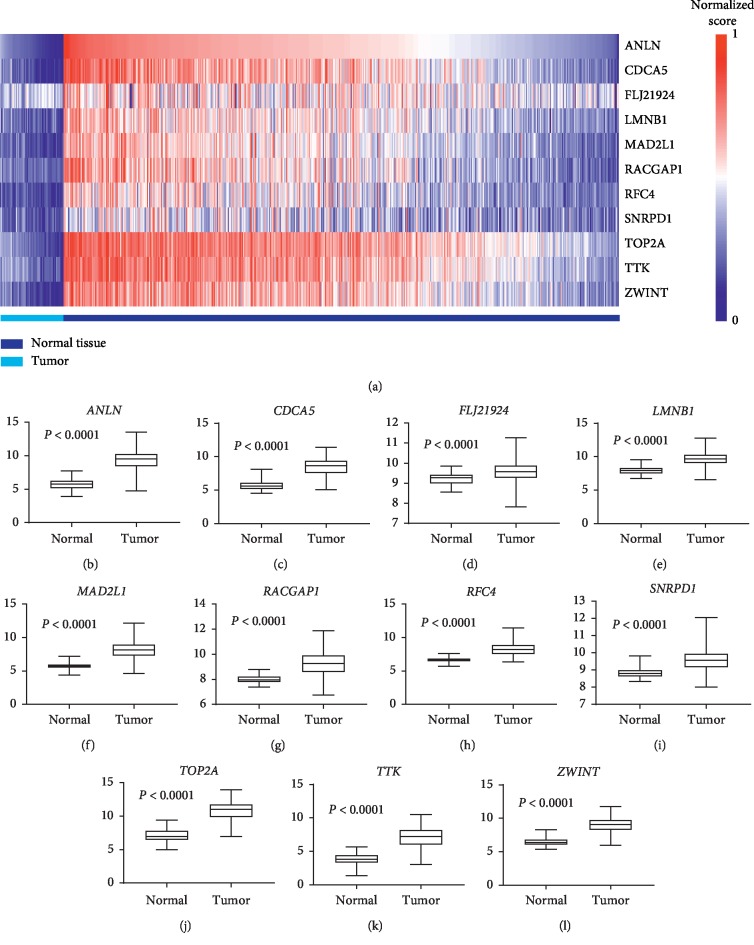
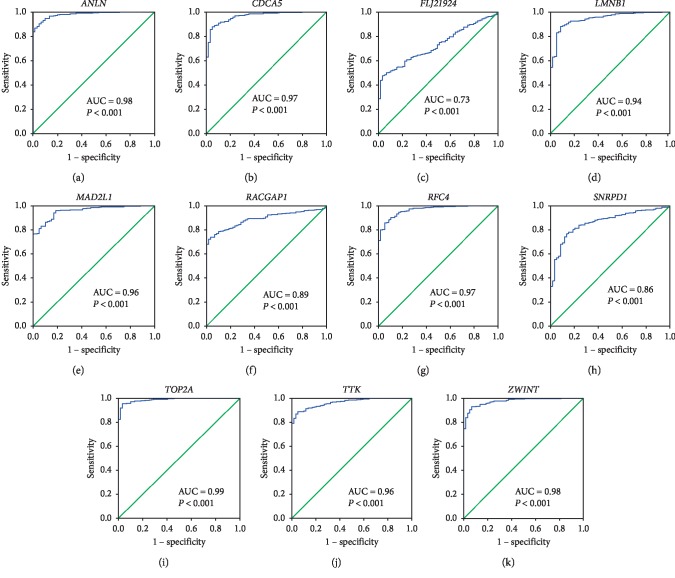
Lung adenocarcinoma is the most frequently diagnosed subtype of nonsmall cell lung cancer. The molecular mechanisms of the initiation and progression of lung adenocarcinoma remain to be further determined. This study aimed to screen genes related to the progression of lung adenocarcinoma. By weighted gene coexpression network analysis (WGCNA), we constructed a free-scale gene coexpression network to evaluate the correlations between multiple gene sets and patients' clinical traits, then further identify predictive biomarkers. GSE11969 was obtained from the Gene Expression Omnibus (GEO) database which contained the gene expression data of 90 lung adenocarcinoma patients. Data of the Cancer Genome Atlas (TCGA) were employed as the validation cohort. After the average linkage hierarchical clustering, a total of 9 modules were generated. In the clinical significant module ( = 0.44, < 0.0001), we identified 29 network hub genes. Subsequent verification in the TCGA database showed that 11 hub genes (, , , , , , , , , , and ) were significantly associated with poor survival data of lung adenocarcinomas. Besides, the results of receiver operating characteristic curves indicated that the mRNA levels of this group of genes exhibited high specificity and sensitivity to distinguish malignant lesions from nonmalignant tissues. Apart from mRNA levels, we found that the protein abundances of these 11 genes were remarkably upregulated in lung adenocarcinomas compared with normal tissues. In conclusion, by the WGCNA method, a panel of 11 genes were identified as predictive biomarkers for tumorigenesis and poor prognosis of lung adenocarcinomas.
肺腺癌是非小细胞肺癌中最常见的亚型。肺腺癌的发生和发展的分子机制仍有待进一步确定。本研究旨在筛选与肺腺癌进展相关的基因。通过加权基因共表达网络分析(WGCNA),我们构建了一个无尺度基因共表达网络,以评估多个基因集与患者临床特征之间的相关性,进而鉴定预测性生物标志物。GSE11969 从基因表达综合数据库(GEO)数据库中获取,该数据库包含 90 例肺腺癌患者的基因表达数据。癌症基因组图谱(TCGA)的数据被用作验证队列。经过平均链接层次聚类,共生成了 9 个模块。在临床显著模块( = 0.44, < 0.0001)中,我们鉴定出 29 个网络枢纽基因。在 TCGA 数据库中的后续验证表明,这 11 个枢纽基因(,,,,,,,,,, and )与肺腺癌患者的不良生存数据显著相关。此外,ROC 曲线的结果表明,这组基因的 mRNA 水平对区分恶性病变与非恶性组织具有较高的特异性和敏感性。除了 mRNA 水平外,我们发现这 11 个基因的蛋白丰度在肺腺癌中明显高于正常组织。总之,通过 WGCNA 方法,鉴定出一组 11 个基因作为肺腺癌发生和预后不良的预测性生物标志物。