Isler Jasmine, Rüfenacht Véronique, Gemperle Corinne, Allegri Gabriella, Häberle Johannes
Division of Metabolism and Children's Research Center University Children's Hospital Zurich Zurich Switzerland.
JIMD Rep. 2020 Jan 9;52(1):28-34. doi: 10.1002/jmd2.12091. eCollection 2020 Mar.
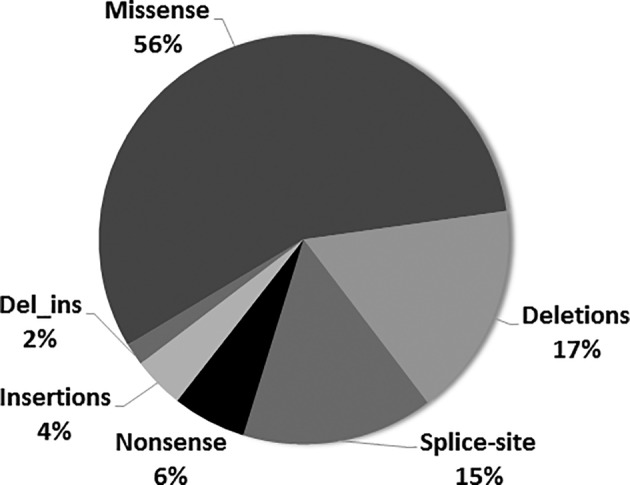
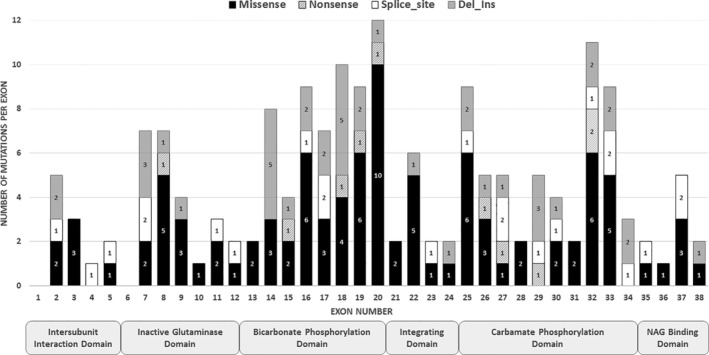
Carbamoylphosphate synthetase 1 (CPS1) deficiency is a rare inborn error of metabolism leading often to neonatal onset hyperammonemia with coma and high mortality. The biochemical features of the disease are nonspecific and cannot distinguish this condition from other defects of the urea cycle, namely -acetylglutamate synthase deficiency. Therefore, molecular genetic investigation is required for confirmation of the disease, and nowadays this is done with increasing frequency applying next-generation sequencing (NGS) techniques. Our laboratory has a long-standing interest in molecular genetic investigation and receives samples from centers in Europe and many other countries. We perform RNA-based molecular genetic investigation as first line investigation and wanted in this study to evaluate our experience with this approach as compared to NGS. In the past 15 years, 297 samples were analyzed, which were referred from 37 countries. CPS1 deficiency could be confirmed in 155 patients carrying 136 different genotypes with only a single mutation recurring more than two times. About 10% of the total 172 variants comprised complex changes (eg, intronic changes possibly affecting splicing, deletions, insertions, or deletions_insertions), which would have been partly missed if only NGS was done. Likewise, RNA analysis was crucial for correct interpretation of at least half of the complex mutations. This study gives highest sensitivity to RNA-based molecular genetic investigation and underlines that NGS should be done together with copy number variation analysis. We propose that unclear cases should be investigated by RNA sequencing in addition, if this method is not used as the initial diagnostic procedure.
氨甲酰磷酸合成酶1(CPS1)缺乏症是一种罕见的先天性代谢缺陷病,常导致新生儿期发作的高氨血症,伴有昏迷,死亡率高。该疾病的生化特征不具有特异性,无法将这种情况与尿素循环的其他缺陷,即N - 乙酰谷氨酸合成酶缺乏症区分开来。因此,需要进行分子遗传学研究来确诊该疾病,如今,应用下一代测序(NGS)技术进行此项研究的频率越来越高。我们实验室长期以来一直对分子遗传学研究感兴趣,并接收来自欧洲和许多其他国家中心的样本。我们将基于RNA的分子遗传学研究作为一线研究方法,并且在本研究中希望评估与NGS相比,我们采用这种方法的经验。在过去15年中,共分析了来自37个国家的297份样本。在155例患者中确诊为CPS1缺乏症,这些患者携带136种不同的基因型,只有一种突变重复出现两次以上。在总共172个变异中,约10%包含复杂变化(例如,可能影响剪接的内含子变化、缺失、插入或缺失插入),如果仅进行NGS,这些变化可能会部分遗漏。同样,RNA分析对于至少一半复杂突变的正确解读至关重要。本研究表明基于RNA的分子遗传学研究具有最高的灵敏度,并强调NGS应与拷贝数变异分析一起进行。我们建议,如果未将RNA测序用作初始诊断程序,对于不明原因的病例应另外进行RNA测序研究。