Children's Medical Center, Peking University First Hospital, Beijing, 102600, China.
Department of Pediatrics, Women and Children's Hospital, School of Medicine, Xiamen University, Xiamen, 361003, China.
BMC Pediatr. 2024 Aug 22;24(1):539. doi: 10.1186/s12887-024-05005-5.
Carbamoyl phosphate synthetase 1 (CPS1) deficiency (OMIM 237300), an autosomal recessive rare and severe urea cycle disorder, is associated with hyperammonemia and high mortality.
Herein we present 12 genetic variants identified in seven clinically well-characterized Chinese patients with CPS1 deficiency who were admitted to the Children's Medical Center of Peking University First Hospital from September 2014 to August 2023.
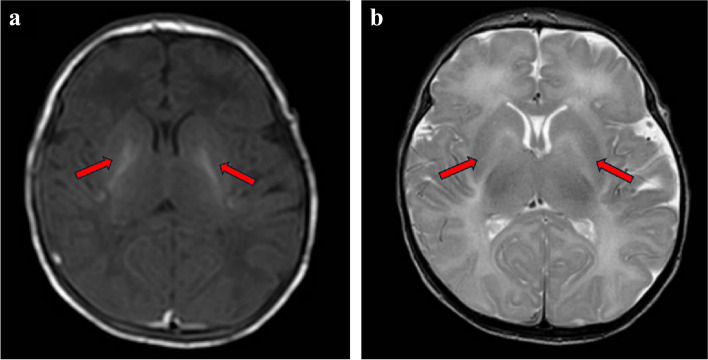
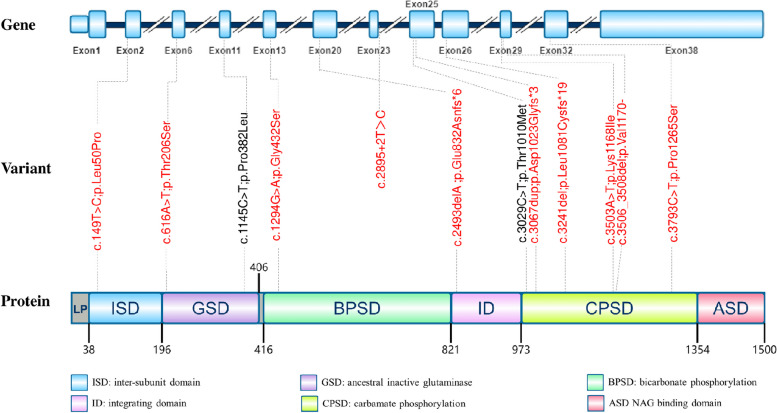
Seven patients (two male and five female patients including two sisters) experienced symptoms onset between 2 days and 13 years of age, and they were diagnosed with CPS1 deficiency between 2 months and 20 years. Peak blood ammonia levels ranged from 160 to 1,000 µmol/L. Three patients showed early-onset CPS1 deficiency, with only one surviving after treatment with sodium phenylbutyrate, N-carbamoyl-L-glutamate, and liver transplantation at 4 months, showing a favorable outcome. The remaining four patients had late-onset CPS1 deficiency, presenting with mental retardation, psychiatric symptoms, and self-selected low-protein diets. Among the 12 CPS1 variants identified in these patients, 10 were novel, with all patients exhibiting compound heterozygosity for CPS1 mutant alleles. Seven variants (c.149T > C, c.616 A > T, c.1145 C > T, c.1294G > A, c.3029 C > T, c.3503 A > T, and c.3793 C > T) resulted in single amino acid substitutions. Three frameshift variations (c.2493del, c.3067dup, and c.3241del) were identified, leading to enzyme truncation. One mutation (c.3506_3508del) caused an in-frame single amino acid deletion, while another (c.2895 + 2T > C) resulted in aberrant splicing.
Except for two known variants, all other variants were identified as novel. No hotspot variants were observed among the patients. Our data contribute to expanding the mutation spectrum of CPS1.
氨甲酰磷酸合成酶 1 缺乏症(CPS1)(OMIM 237300)是一种常染色体隐性遗传的罕见且严重的尿素循环障碍,与高血氨和高死亡率有关。
本文报告了 7 例临床特征明确的 CPS1 缺乏症患者中发现的 12 种遗传变异,这些患者于 2014 年 9 月至 2023 年 8 月期间在北京大学第一医院儿童医学中心就诊。
7 例患者(2 例男性和 5 例女性,包括 2 例姐妹)发病年龄为 2 天至 13 岁,发病后 2 个月至 20 年间确诊为 CPS1 缺乏症。峰值血氨水平为 160 至 1000 μmol/L。3 例为早发型 CPS1 缺乏症,其中仅 1 例在 4 个月时接受苯丁酸钠、N-氨甲酰-L-谷氨酸和肝移植治疗后存活,预后良好。其余 4 例为晚发型 CPS1 缺乏症,表现为智力障碍、精神症状和自行选择低蛋白饮食。在这些患者中发现的 12 种 CPS1 变异中,有 10 种为新发现的变异,所有患者均表现为 CPS1 突变等位基因的复合杂合性。7 种变异(c.149T>C、c.616A>T、c.1145C>T、c.1294G>A、c.3029C>T、c.3503A>T 和 c.3793C>T)导致单个氨基酸取代。发现 3 种移码变异(c.2493del、c.3067dup 和 c.3241del),导致酶截短。1 种突变(c.3506_3508del)导致框内单个氨基酸缺失,另 1 种突变(c.2895+2T>C)导致异常剪接。
除了两种已知的变异,其余变异均为新发现的变异。患者中未发现热点变异。本研究结果有助于扩展 CPS1 的突变谱。