Hui Jialiang, Liu Ruiyu, Zhang Haibo, He Shuhua, Wei Anyang
Department of Urology, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong, China.
PeerJ. 2020 Feb 28;8:e8653. doi: 10.7717/peerj.8653. eCollection 2020.
Erectile dysfunction (ED) is one of the most common male-disease globally. Despite efforts to explain its pathogenesis, the molecular mechanisms of ED are still not well understood.
The microarray dataset GSE10804 was downloaded from the Gene Expression Omnibus (GEO) to find candidate genes in ED progression. After differentially expressed genes (DEGs) were identified, functional enrichment analysis was performed. In addition, a protein-protein interaction network (PPI) was established and module analysis was performed through the STRING and Cytoscape.
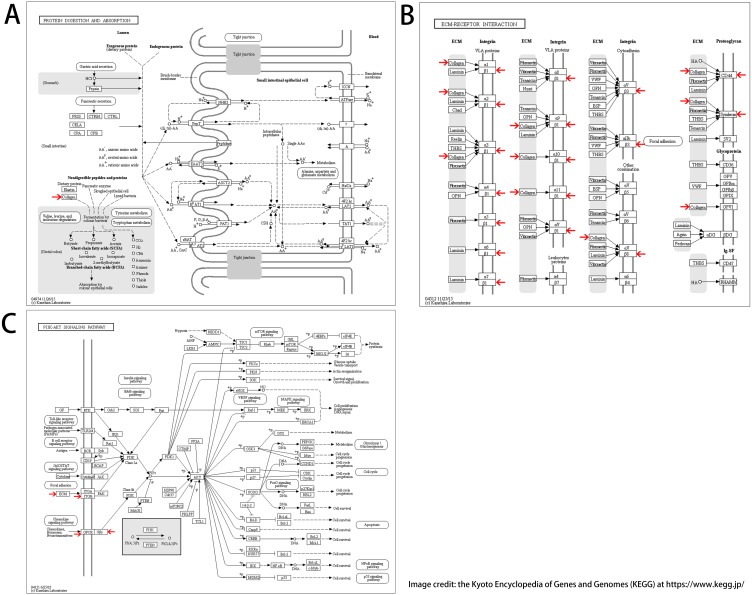
A total of 618 DEGs were identified in all, containing 430 downregulated genes and 188 upregulated genes. The enriched functions and pathways of the DEGs include transcription from RNA polymerase II promoter, cell adhesion, calcium ion binding, receptor binding, Akt signaling pathway, receptor interaction, protein digestion, and absorption. We picked out twenty-five hub genes, with biological process (BP) analyses revealing that the genes were principally associated with cellular responses to amino acid stimuli, extracellular matrix structural constituent, collagen trimer, protein digestion and absorption, ECM-receptor interaction and PI3K-Akt signaling pathway. To sum up, DEGs and hub genes distinguished in this study not only help us understand the molecular mechanisms behind the carcinogenesis and progression of ED, but also play a part in the diagnosis and treatment of ED by providing candidate targets.
勃起功能障碍(ED)是全球最常见的男性疾病之一。尽管人们努力解释其发病机制,但ED的分子机制仍未得到充分理解。
从基因表达综合数据库(GEO)下载微阵列数据集GSE10804,以寻找ED进展中的候选基因。在鉴定出差异表达基因(DEG)后,进行功能富集分析。此外,建立了蛋白质-蛋白质相互作用网络(PPI),并通过STRING和Cytoscape进行模块分析。
总共鉴定出618个DEG,其中包括430个下调基因和188个上调基因。DEG的富集功能和途径包括RNA聚合酶II启动子转录、细胞粘附、钙离子结合、受体结合、Akt信号通路、受体相互作用、蛋白质消化和吸收。我们挑选出25个枢纽基因,生物学过程(BP)分析表明这些基因主要与细胞对氨基酸刺激的反应、细胞外基质结构成分、胶原三聚体、蛋白质消化和吸收、ECM-受体相互作用以及PI3K-Akt信号通路有关。综上所述,本研究中鉴定出的DEG和枢纽基因不仅有助于我们理解ED发生和进展背后的分子机制,还通过提供候选靶点在ED的诊断和治疗中发挥作用。