Cardiology Division, Department of Medicine, The University of Hong Kong, Hong Kong SAR, China.
Hong Kong-Guangdong Joint Laboratory on Stem Cell and Regenerative Medicine, The University of Hong Kong, Hong Kong SAR, China.
Cardiovasc Res. 2021 Feb 22;117(3):767-779. doi: 10.1093/cvr/cvaa019.
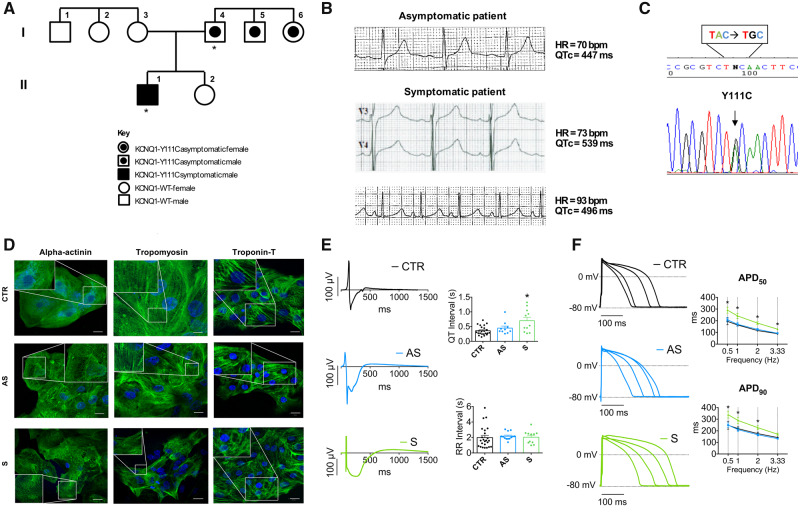
In long QT syndrome (LQTS) patients, modifier genes modulate the arrhythmic risk associated with a disease-causing mutation. Their recognition can improve risk stratification and clinical management, but their discovery represents a challenge. We tested whether a cellular-driven approach could help to identify new modifier genes and especially their mechanism of action.
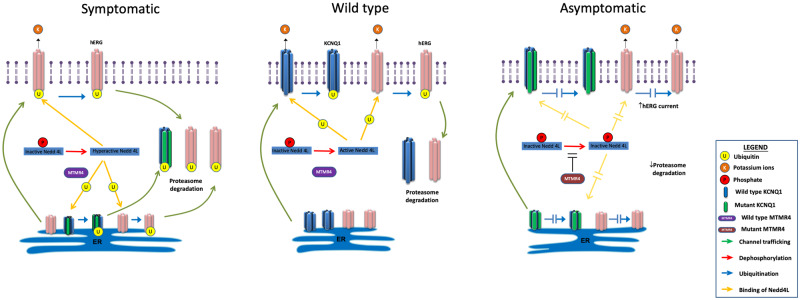
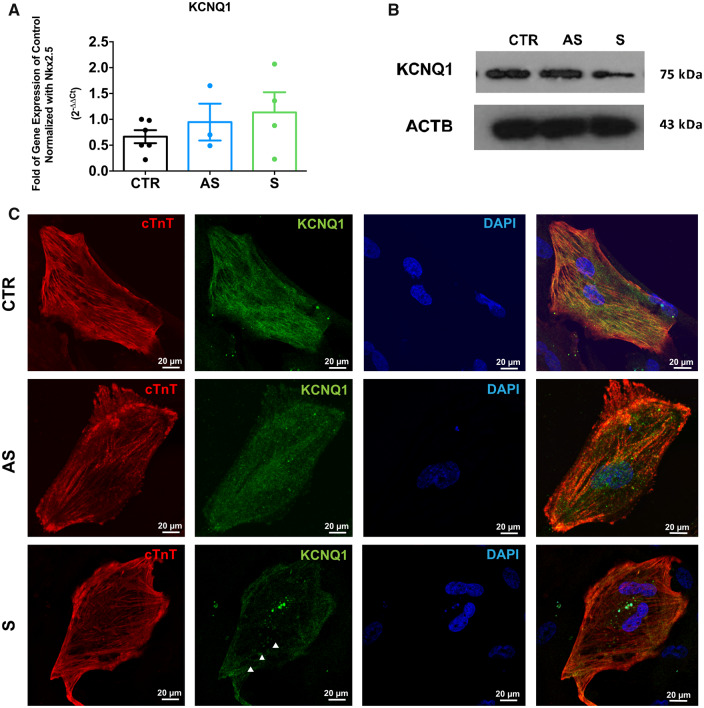
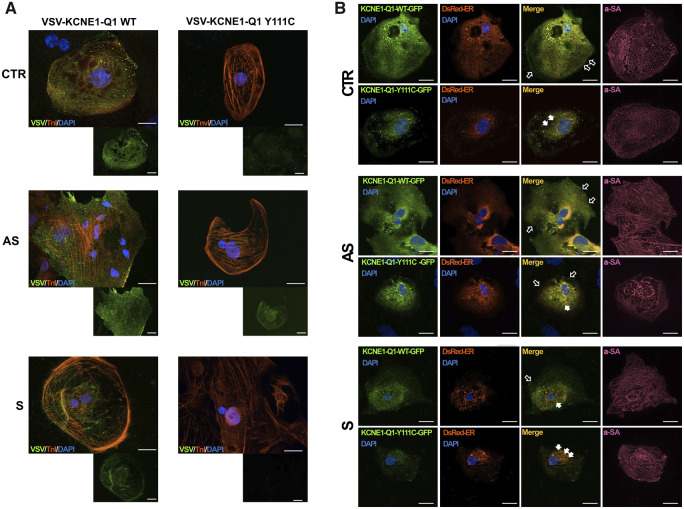
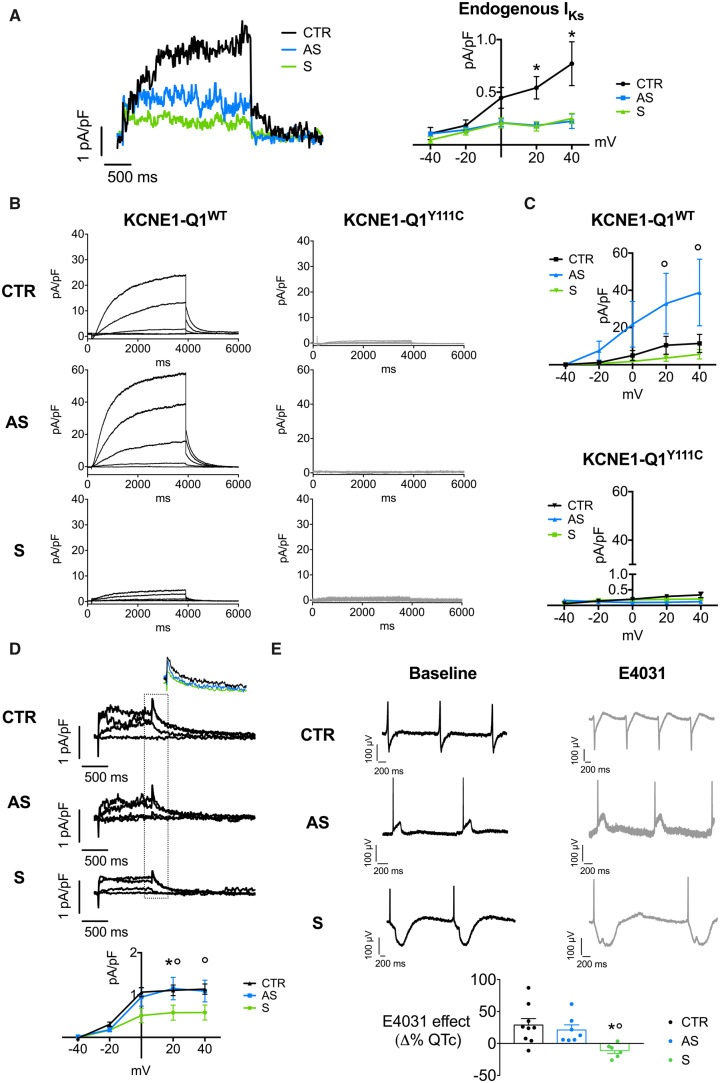
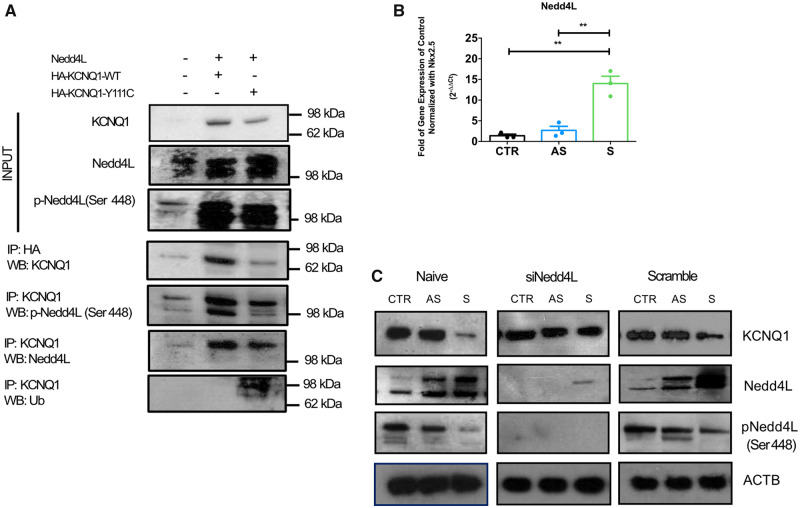
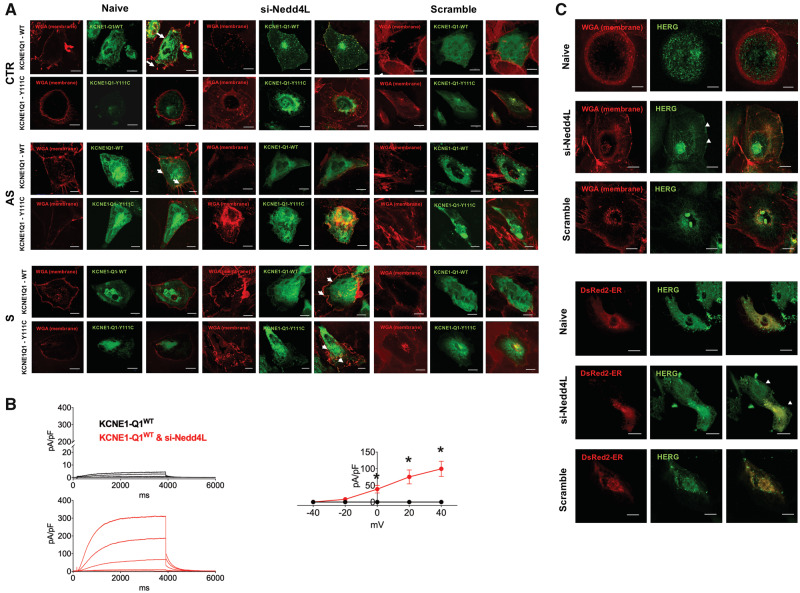
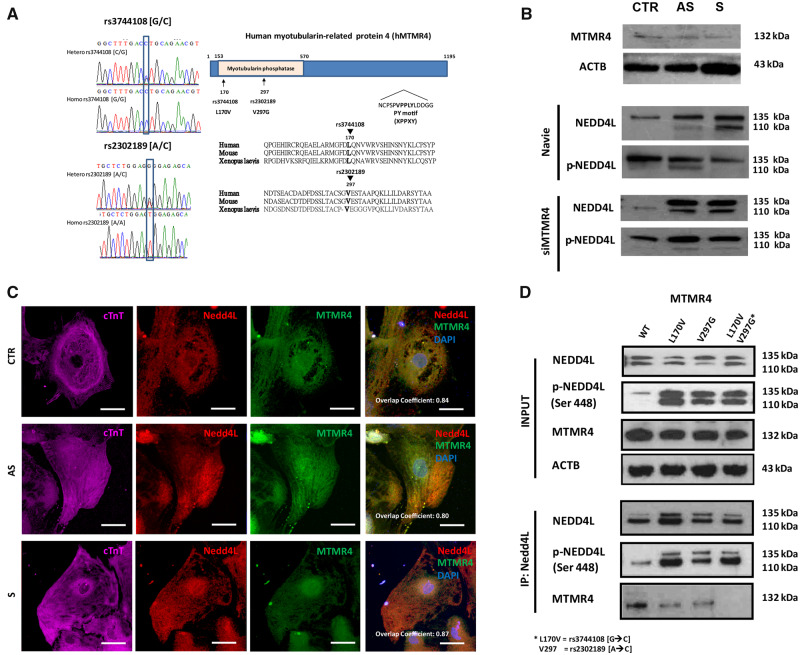
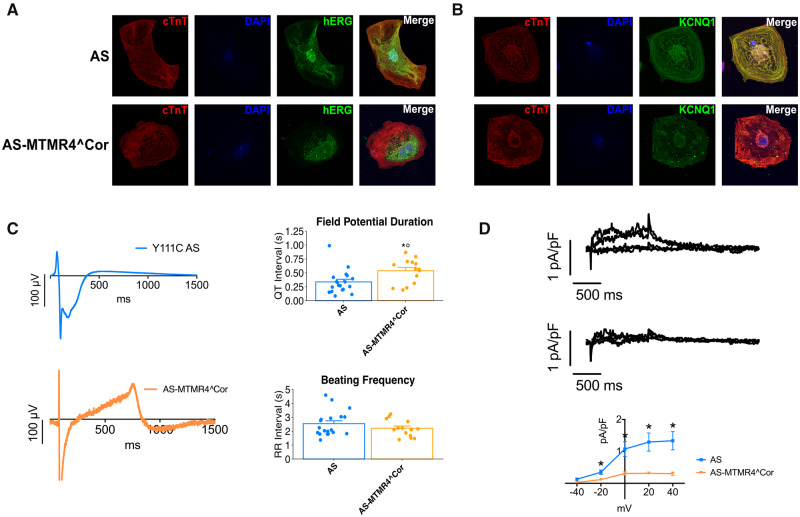
We generated human-induced pluripotent stem cell-derived cardiomyocytes (iPSC-CM) from two patients carrying the same KCNQ1-Y111C mutation, but presenting opposite clinical phenotypes. We showed that the phenotype of the iPSC-CMs derived from the symptomatic patient is due to impaired trafficking and increased degradation of the mutant KCNQ1 and wild-type human ether-a-go-go-related gene. In the iPSC-CMs of the asymptomatic (AS) patient, the activity of an E3 ubiquitin-protein ligase (Nedd4L) involved in channel protein degradation was reduced and resulted in a decreased arrhythmogenic substrate. Two single-nucleotide variants (SNVs) on the Myotubularin-related protein 4 (MTMR4) gene, an interactor of Nedd4L, were identified by whole-exome sequencing as potential contributors to decreased Nedd4L activity. Correction of these SNVs by CRISPR/Cas9 unmasked the LQTS phenotype in AS cells. Importantly, the same MTMR4 variants were present in 77% of AS Y111C mutation carriers of a separate cohort. Thus, genetically mediated interference with Nedd4L activation seems associated with protective effects.
Our finding represents the first demonstration of the cellular mechanism of action of a protective modifier gene in LQTS. It provides new clues for advanced risk stratification and paves the way for the design of new therapies targeting this specific molecular pathway.
在长 QT 综合征 (LQTS) 患者中,修饰基因调节与致病突变相关的心律失常风险。识别它们可以改善风险分层和临床管理,但发现它们是一个挑战。我们测试了一种细胞驱动的方法是否有助于识别新的修饰基因,特别是它们的作用机制。
我们从携带相同 KCNQ1-Y111C 突变但表现出相反临床表型的两名患者中生成了人诱导多能干细胞衍生的心肌细胞 (iPSC-CM)。我们表明,来自有症状患者的 iPSC-CM 的表型是由于突变的 KCNQ1 和野生型人类 ether-a-go-go 相关基因的转运受损和降解增加所致。在无症状 (AS) 患者的 iPSC-CMs 中,参与通道蛋白降解的 E3 泛素蛋白连接酶 (Nedd4L) 的活性降低,导致心律失常底物减少。通过全外显子组测序,在 Nedd4L 的相互作用蛋白肌管相关蛋白 4 (MTMR4) 基因上发现了两个单核苷酸变异 (SNV),它们可能导致 Nedd4L 活性降低。通过 CRISPR/Cas9 对这些 SNV 进行校正,揭示了 AS 细胞中的 LQTS 表型。重要的是,在一个单独队列中,77%的 AS Y111C 突变携带者存在相同的 MTMR4 变体。因此,遗传介导的对 Nedd4L 激活的干扰似乎与保护作用相关。
我们的发现代表了在 LQTS 中修饰基因作用机制的首次证明。它为高级风险分层提供了新的线索,并为针对该特定分子途径的新疗法的设计铺平了道路。