Department of Chemistry, Roger Adams Laboratory, University of Illinois, Urbana, IL, USA.
Pfizer Worldwide Research and Development, Groton Laboratories, Groton, CT, USA.
Nature. 2020 Apr;580(7805):621-627. doi: 10.1038/s41586-020-2137-8. Epub 2020 Mar 16.
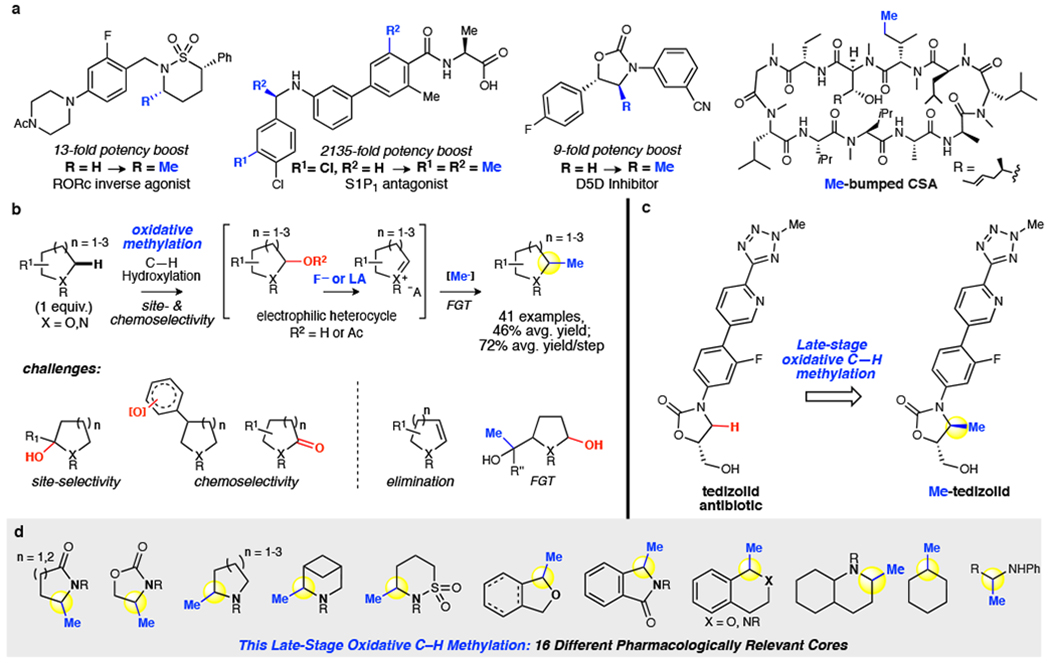
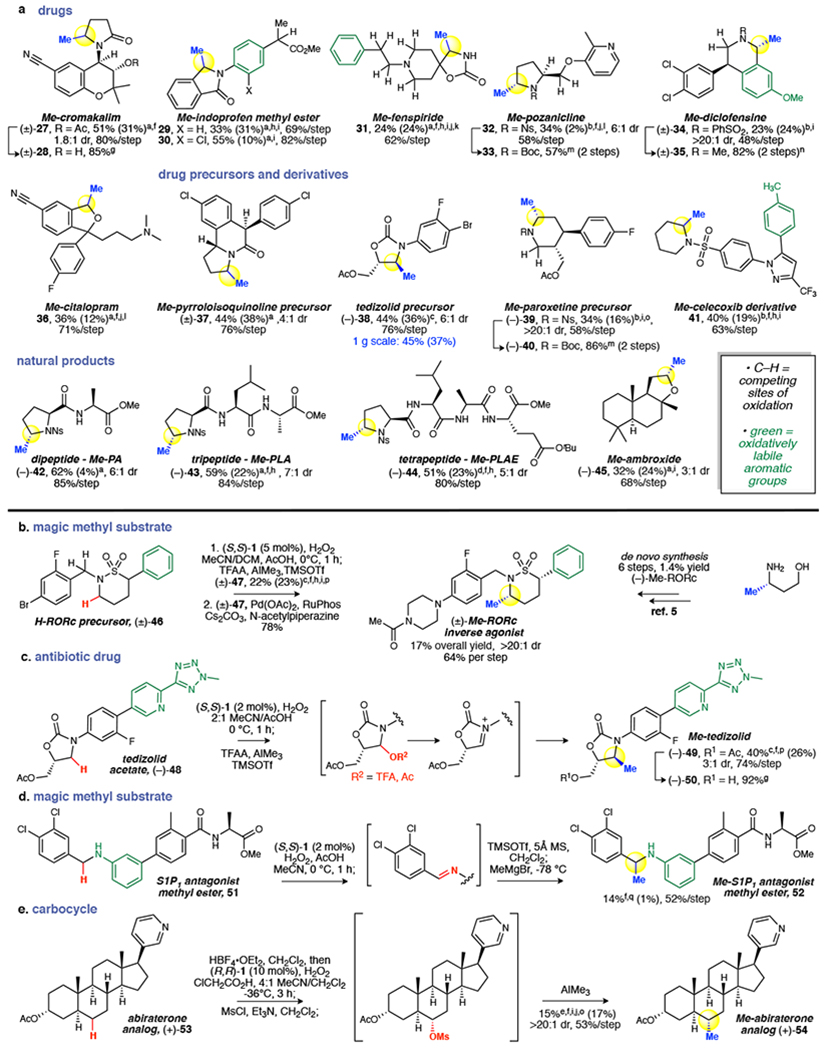
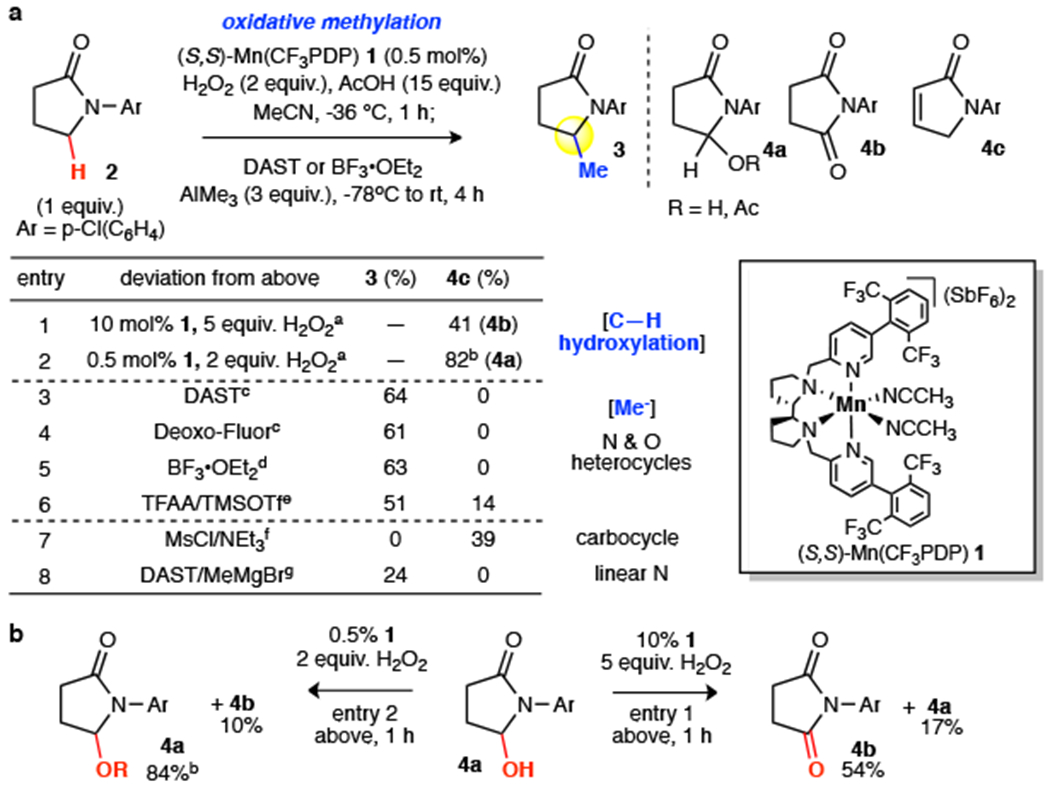
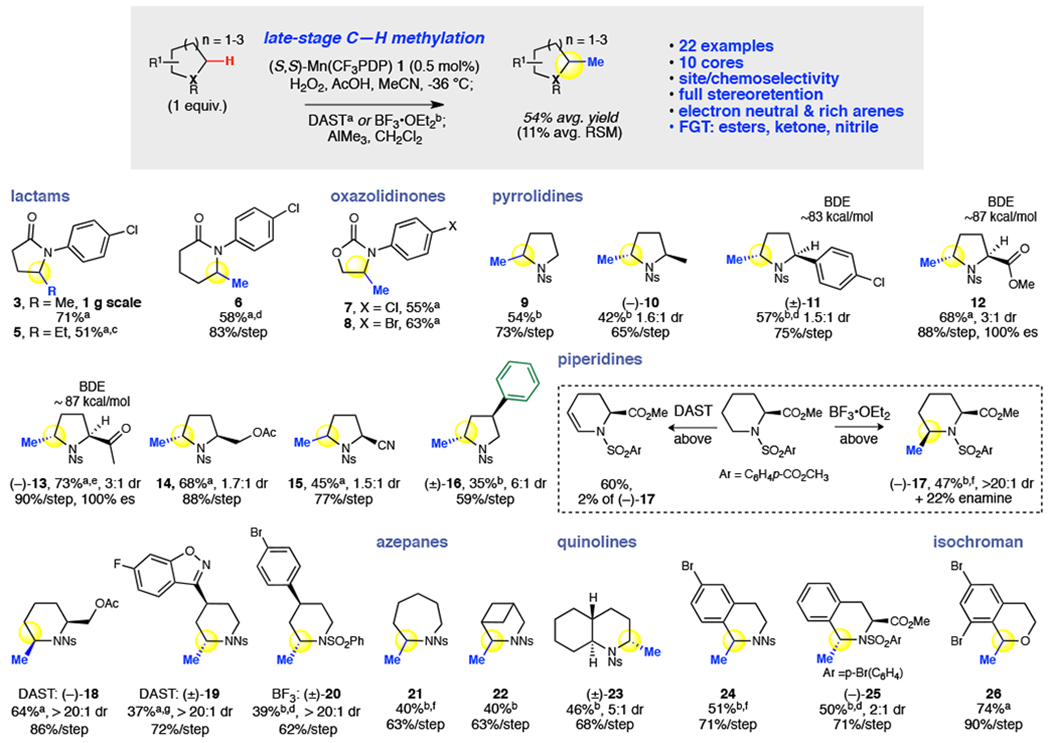
Frequently referred to as the 'magic methyl effect', the installation of methyl groups-especially adjacent (α) to heteroatoms-has been shown to dramatically increase the potency of biologically active molecules. However, existing methylation methods show limited scope and have not been demonstrated in complex settings. Here we report a regioselective and chemoselective oxidative C(sp)-H methylation method that is compatible with late-stage functionalization of drug scaffolds and natural products. This combines a highly site-selective and chemoselective C-H hydroxylation with a mild, functional-group-tolerant methylation. Using a small-molecule manganese catalyst, Mn(CFPDP), at low loading (at a substrate/catalyst ratio of 200) affords targeted C-H hydroxylation on heterocyclic cores, while preserving electron-neutral and electron-rich aryls. Fluorine- or Lewis-acid-assisted formation of reactive iminium or oxonium intermediates enables the use of a mildly nucleophilic organoaluminium methylating reagent that preserves other electrophilic functionalities on the substrate. We show this late-stage C(sp)-H methylation on 41 substrates housing 16 different medicinally important cores that include electron-rich aryls, heterocycles, carbonyls and amines. Eighteen pharmacologically relevant molecules with competing sites-including drugs (for example, tedizolid) and natural products-are methylated site-selectively at the most electron rich, least sterically hindered position. We demonstrate the syntheses of two magic methyl substrates-an inverse agonist for the nuclear receptor RORc and an antagonist of the sphingosine-1-phosphate receptor-1-via late-stage methylation from the drug or its advanced precursor. We also show a remote methylation of the B-ring carbocycle of an abiraterone analogue. The ability to methylate such complex molecules at late stages will reduce synthetic effort and thereby expedite broader exploration of the magic methyl effect in pursuit of new small-molecule therapeutics and chemical probes.
经常被称为“神奇甲基效应”,安装甲基基团 - 特别是相邻的(α)杂原子 - 已被证明可以显著提高生物活性分子的效力。然而,现有的甲基化方法显示出有限的范围,并且尚未在复杂环境中得到证明。在这里,我们报告了一种区域选择性和化学选择性的氧化 C(sp)-H 甲基化方法,该方法与药物支架和天然产物的后期功能化兼容。这结合了高度选择性和化学选择性的 C-H 羟化与温和的、官能团耐受的甲基化。使用低负载量(在底物/催化剂比为 200 的情况下)的小分子锰催化剂 Mn(CFPDP),可以在杂环核心上进行靶向 C-H 羟化,同时保留电子中性和富电子芳基。氟或路易斯酸辅助形成反应性亚铵或氧鎓中间体,可使用温和的亲核有机铝甲基化试剂,同时保留底物上的其他亲电官能团。我们展示了在 41 个含有 16 种不同药用核心的底物上的晚期 C(sp)-H 甲基化,这些核心包括富电子芳基、杂环、羰基和胺。在包括药物(例如 tedizolid)和天然产物在内的 18 种具有竞争位点的药理相关分子中,十八种具有竞争位点的药物(例如 tedizolid)和天然产物)在最富电子、最少空间位阻的位置上进行了位点选择性甲基化。我们通过药物或其高级前体的晚期甲基化,展示了两种神奇甲基底物的合成 - 核受体 RORc 的反向激动剂和鞘氨醇-1-磷酸受体-1 的拮抗剂。我们还展示了 abiraterone 类似物的 B 环碳环的远程甲基化。在晚期对如此复杂的分子进行甲基化的能力将减少合成工作量,从而加快对神奇甲基效应的更广泛探索,以寻求新的小分子治疗剂和化学探针。