Gene Therapy Program, University of Pennsylvania Perelman School of Medicine, 125 South 31st Street, Suite 1200, Philadelphia, PA, 19104, USA.
BMC Genomics. 2020 Mar 17;21(1):239. doi: 10.1186/s12864-020-6655-4.
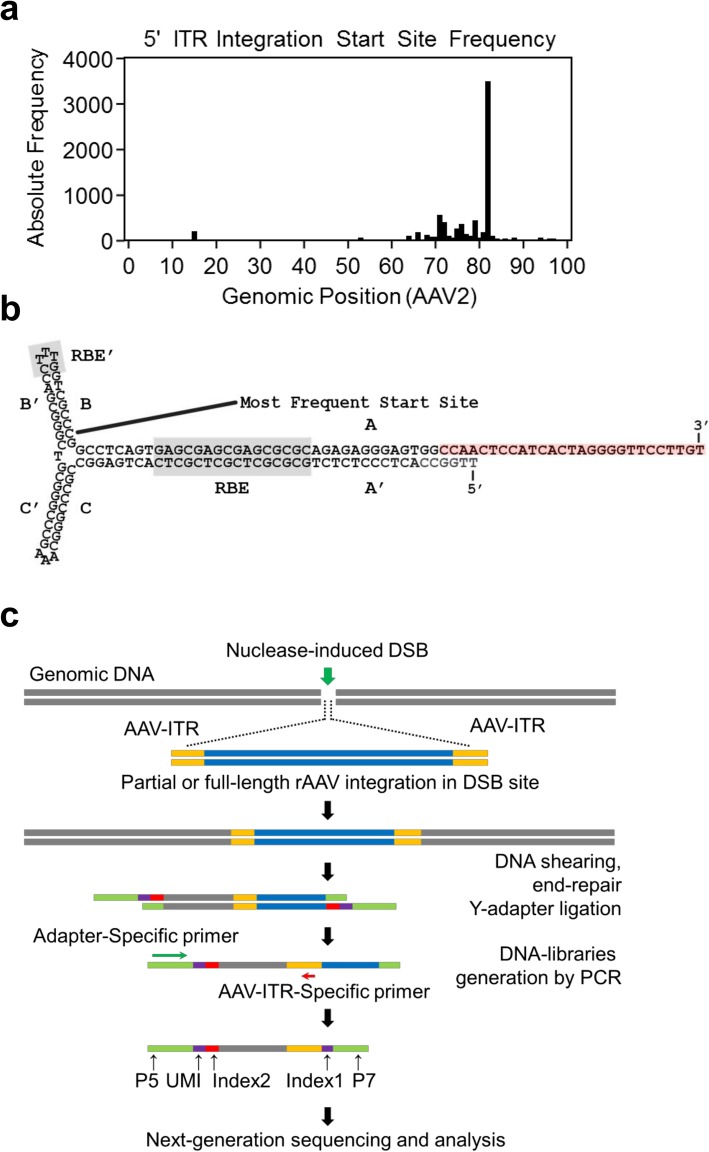
Identifying nuclease-induced double-stranded breaks in DNA on a genome-wide scale is critical for assessing the safety and efficacy of genome editing therapies. We previously demonstrated that after administering adeno-associated viral (AAV) vector-mediated genome-editing strategies in vivo, vector sequences integrated into the host organism's genomic DNA at double-stranded breaks. Thus, identifying the genomic location of inserted AAV sequences would enable us to identify DSB events, mainly derived from the nuclease on- and off-target activity.
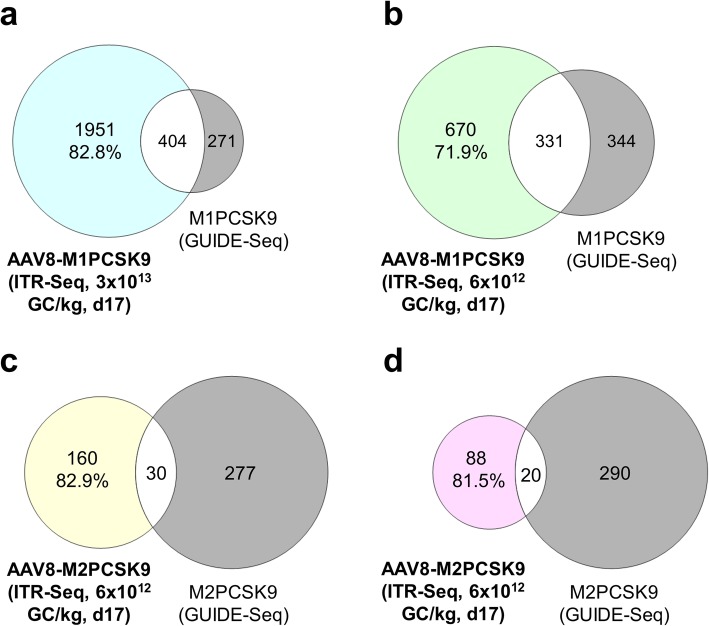
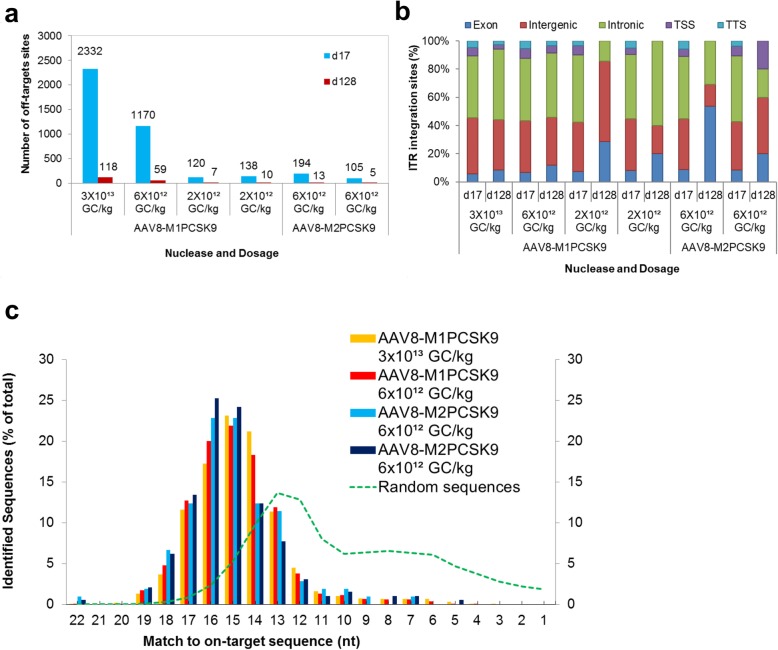
Here, we developed a next-generation sequencing assay that detects insertions of specific AAV vector sequences called inverted terminal repeats (ITRs). This assay, ITR-Seq, enables us to identify off-target nuclease activity in vivo. Using ITR-Seq, we analyzed liver DNA samples of rhesus macaques treated with AAV vectors expressing a meganuclease. We found dose-dependent off-target activity and reductions in off-target events induced by further meganuclease development. In mice, we identified the genomic locations of ITR integration after treatment with Cas9 nucleases and their corresponding single-guide RNAs.
In sum, ITR-Seq is a powerful method for identifying off-target sequences induced by AAV vector-delivered genome-editing nucleases. ITR-Seq will help us understand the specificity and efficacy of different genome-editing nucleases in animal models and clinical studies. This information can help enhance the safety profile of gene-editing therapies.
在全基因组范围内识别 DNA 中由核酸酶诱导的双链断裂对于评估基因组编辑疗法的安全性和有效性至关重要。我们之前证明,在体内给予腺相关病毒(AAV)载体介导的基因组编辑策略后,载体序列会在双链断裂处整合到宿主生物的基因组 DNA 中。因此,确定插入 AAV 序列的基因组位置将使我们能够识别 DSB 事件,这些事件主要源自核酸酶的脱靶和靶内活性。
在这里,我们开发了一种能够检测特定 AAV 载体序列(称为反向末端重复序列 [ITR])插入的下一代测序检测方法。该检测方法 ITR-Seq 使我们能够在体内识别脱靶核酸酶活性。使用 ITR-Seq,我们分析了用表达 meganuclease 的 AAV 载体处理的恒河猴肝脏 DNA 样本。我们发现脱靶活性与剂量相关,并且进一步的 meganuclease 开发减少了脱靶事件的发生。在小鼠中,我们在 Cas9 核酸酶及其相应的单链引导 RNA 处理后鉴定了 ITR 整合的基因组位置。
总之,ITR-Seq 是一种用于识别由 AAV 载体递送的基因组编辑核酸酶诱导的脱靶序列的强大方法。ITR-Seq 将帮助我们了解不同基因组编辑核酸酶在动物模型和临床研究中的特异性和功效。这些信息可以帮助提高基因编辑疗法的安全性。