Univ. Lille, Inserm, Institut Pasteur de Lille, U1177 Drugs and Molecules for Living Systems, F-59000 Lille, France.
Brief Bioinform. 2021 Mar 22;22(2):1790-1818. doi: 10.1093/bib/bbaa034.
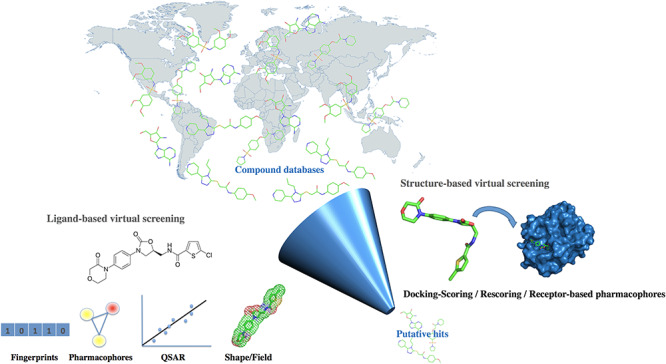
The interplay between life sciences and advancing technology drives a continuous cycle of chemical data growth; these data are most often stored in open or partially open databases. In parallel, many different types of algorithms are being developed to manipulate these chemical objects and associated bioactivity data. Virtual screening methods are among the most popular computational approaches in pharmaceutical research. Today, user-friendly web-based tools are available to help scientists perform virtual screening experiments. This article provides an overview of internet resources enabling and supporting chemical biology and early drug discovery with a main emphasis on web servers dedicated to virtual ligand screening and small-molecule docking. This survey first introduces some key concepts and then presents recent and easily accessible virtual screening and related target-fishing tools as well as briefly discusses case studies enabled by some of these web services. Notwithstanding further improvements, already available web-based tools not only contribute to the design of bioactive molecules and assist drug repositioning but also help to generate new ideas and explore different hypotheses in a timely fashion while contributing to teaching in the field of drug development.
生命科学与先进技术的相互作用推动了化学数据的持续增长;这些数据通常存储在开放或部分开放的数据库中。与此同时,许多不同类型的算法也在被开发出来,以处理这些化学对象和相关的生物活性数据。虚拟筛选方法是药物研究中最受欢迎的计算方法之一。如今,用户友好的基于网络的工具可用于帮助科学家进行虚拟筛选实验。本文概述了支持化学生物学和早期药物发现的互联网资源,主要侧重于专门用于虚拟配体筛选和小分子对接的网络服务器。该调查首先介绍了一些关键概念,然后介绍了最近和易于访问的虚拟筛选和相关的靶标捕捞工具,并简要讨论了其中一些网络服务支持的案例研究。尽管还需要进一步改进,但现有的基于网络的工具不仅有助于设计生物活性分子和辅助药物重定位,还有助于及时产生新的想法和探索不同的假设,同时为药物开发领域的教学做出贡献。