Roberts Wade R, Roalson Eric H
School of Biological Sciences, Washington State University, Pullman, WA, USA.
Biological Sciences, University of Arkansas, Fayetteville, AR, USA.
PeerJ. 2020 Mar 11;8:e8778. doi: 10.7717/peerj.8778. eCollection 2020.
Genetic pathways involved with flower color and shape are thought to play an important role in the development of flowers associated with different pollination syndromes, such as those associated with bee, butterfly, or hummingbird pollination. Because pollination syndromes are complex traits that are orchestrated by multiple genes and pathways, the gene regulatory networks have not been explored. Gene co-expression networks provide a systems level approach to identify important contributors to floral diversification.
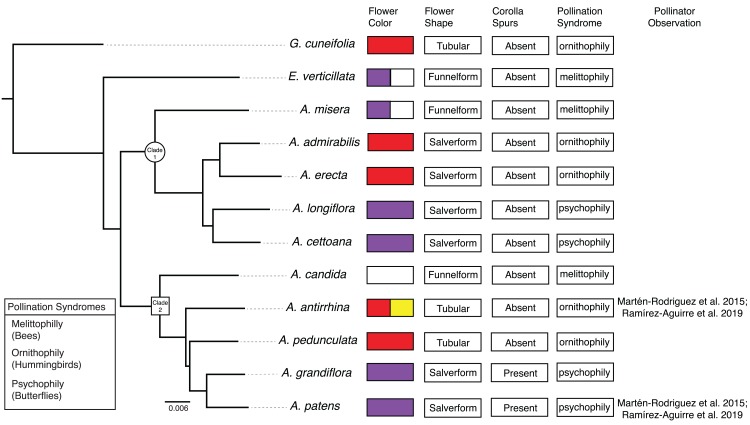
RNA-sequencing was used to assay gene expression across two stages of flower development (an early bud and an intermediate stage) in 10 species of (Gesneriaceae). Two stage-specific co-expression networks were created from 9,503 orthologs and analyzed to identify module hubs and the network periphery. Module association with bee, butterfly, and hummingbird pollination syndromes was tested using phylogenetic mixed models. The relationship between network connectivity and evolutionary rates ( / ) was tested using linear models.
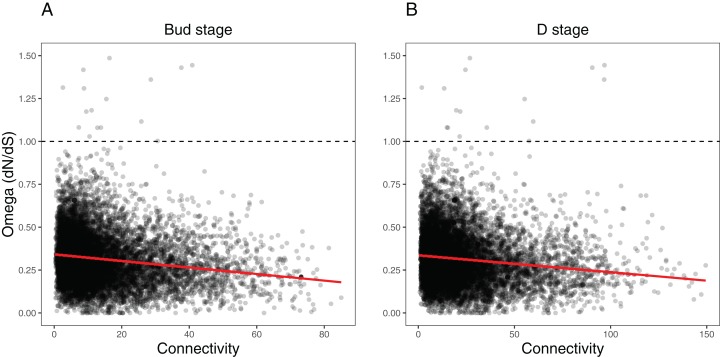
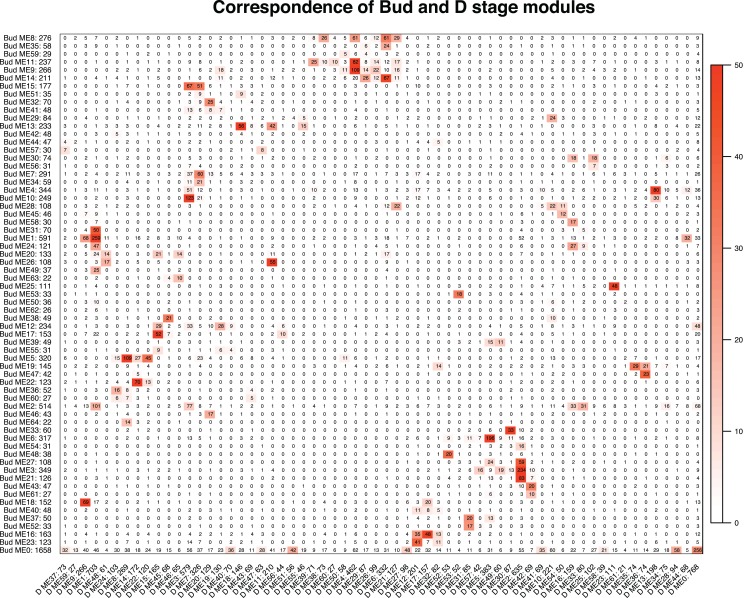
Networks contained 65 and 62 modules that were largely preserved between developmental stages and contained few stage-specific modules. Over a third of the modules in both networks were associated with flower color, shape, and pollination syndrome. Within these modules, several hub nodes were identified that related to the production of anthocyanin and carotenoid pigments and the development of flower shape. Evolutionary rates were decreased in highly connected genes and elevated in peripheral genes.
This study aids in the understanding of the genetic architecture and network properties underlying the development of floral form and provides valuable candidate modules and genes for future studies.
与花色和花形相关的遗传途径被认为在与不同传粉综合征相关的花的发育中起重要作用,例如与蜜蜂、蝴蝶或蜂鸟传粉相关的花。由于传粉综合征是由多个基因和途径协调的复杂性状,基因调控网络尚未得到探索。基因共表达网络提供了一种系统水平的方法来识别花多样化的重要贡献者。
使用RNA测序来分析10种苦苣苔科植物花发育的两个阶段(早期花蕾和中期)的基因表达。从9503个直系同源基因创建了两个阶段特异性共表达网络,并进行分析以识别模块枢纽和网络边缘。使用系统发育混合模型测试模块与蜜蜂、蝴蝶和蜂鸟传粉综合征的关联。使用线性模型测试网络连通性与进化速率(ω/λ)之间的关系。
网络包含65和62个模块,这些模块在发育阶段之间基本保留,并且很少有阶段特异性模块。两个网络中超过三分之一的模块与花色、花形和传粉综合征相关。在这些模块中,鉴定出了几个与花青素和类胡萝卜素色素产生以及花形发育相关的枢纽节点。高度连通的基因进化速率降低,外围基因进化速率升高。
本研究有助于理解花形态发育的遗传结构和网络特性,并为未来研究提供有价值的候选模块和基因。