Zaheed Oza, Samson Julia, Dean Kellie
School of Biochemistry and Cell Biology, Western Gateway Building, University College Cork, Cork, T12XF62, Ireland.
Noncoding RNA Res. 2020 Feb 24;5(2):48-59. doi: 10.1016/j.ncrna.2020.02.004. eCollection 2020 Jun.
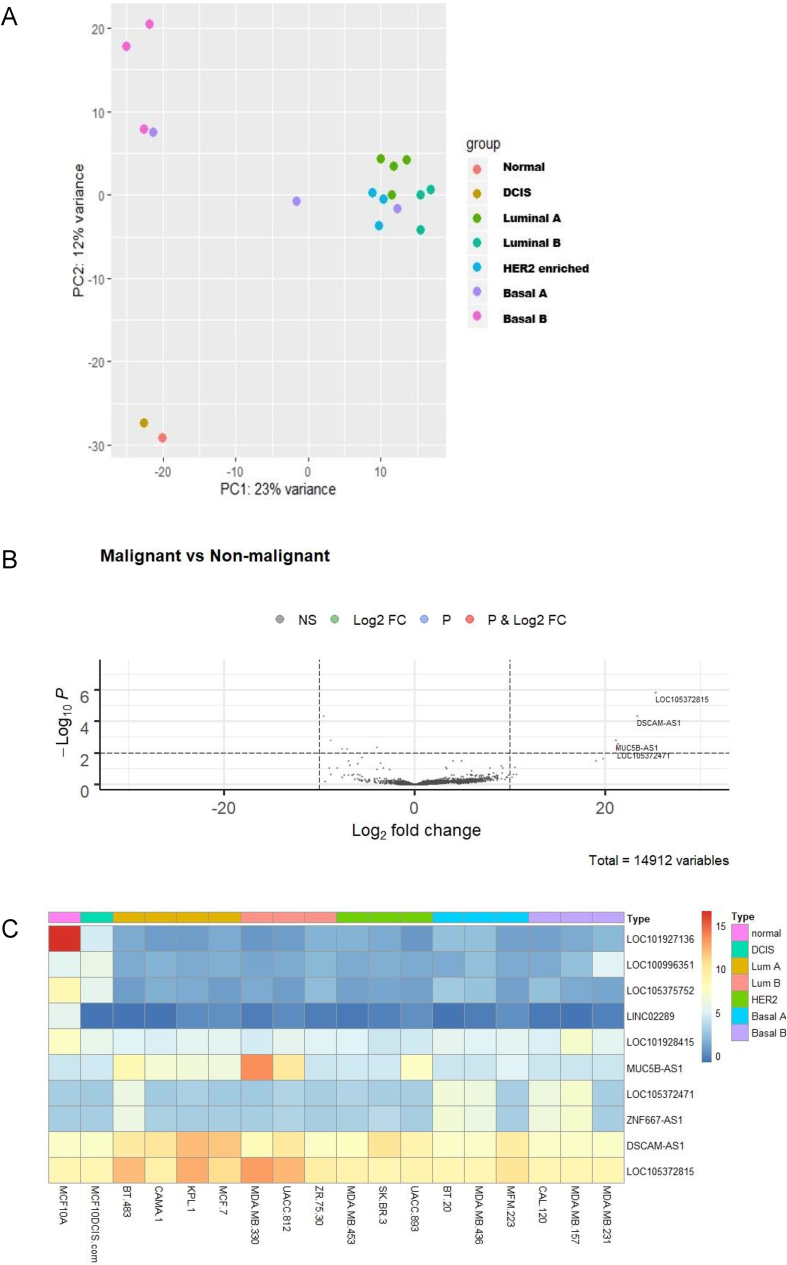
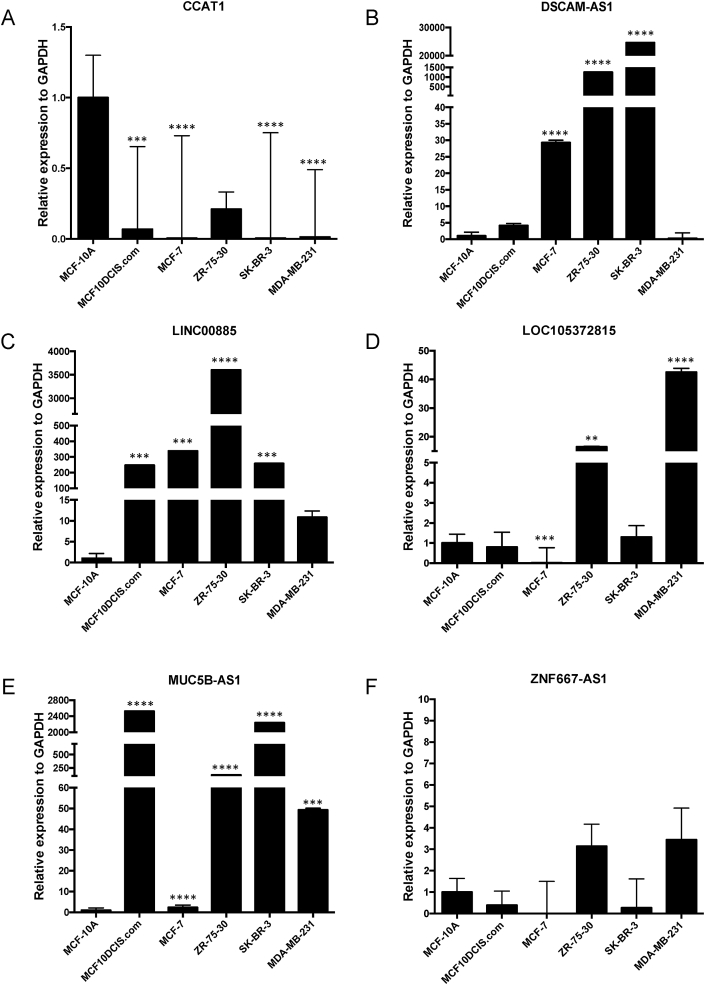
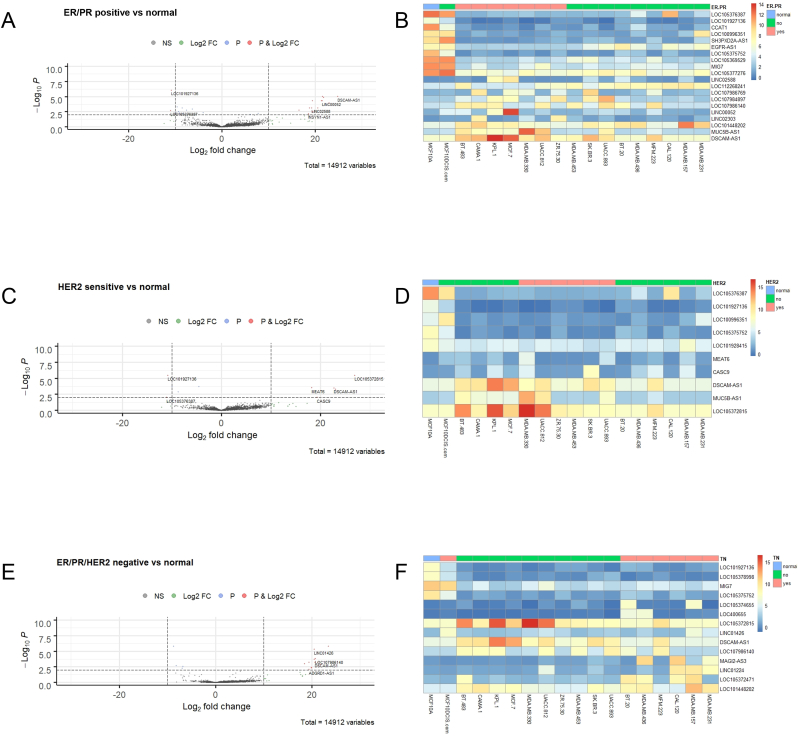
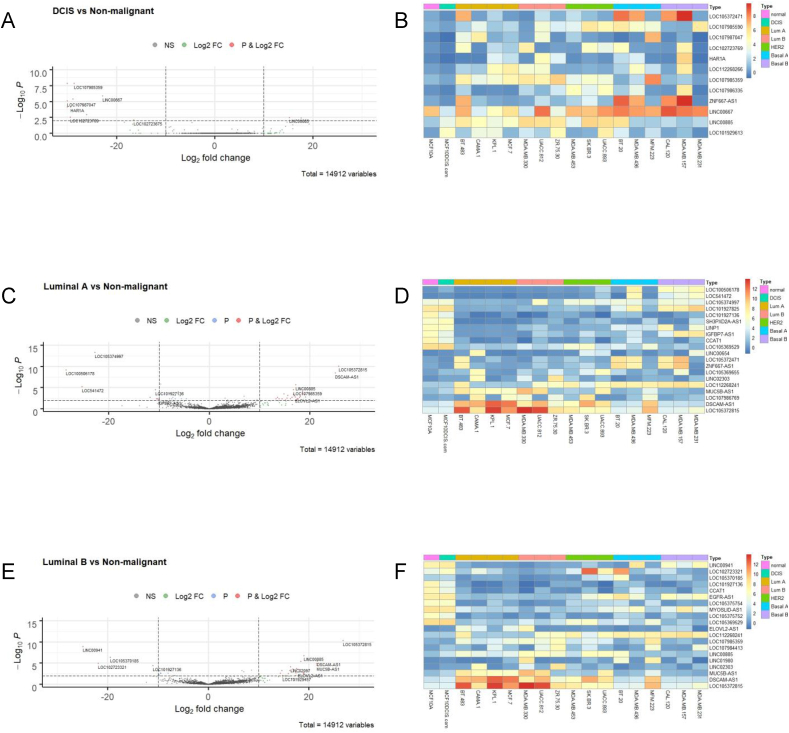
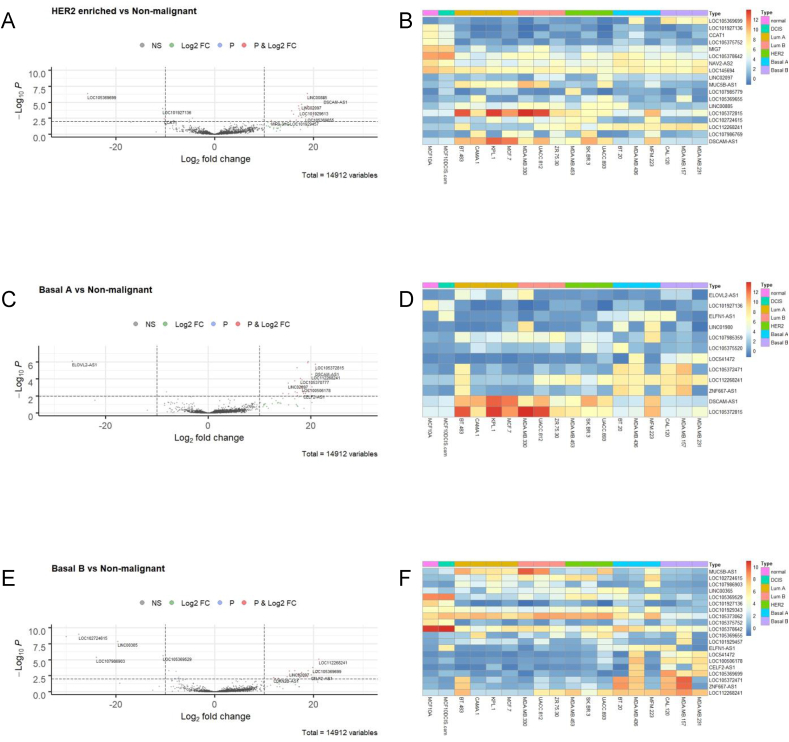
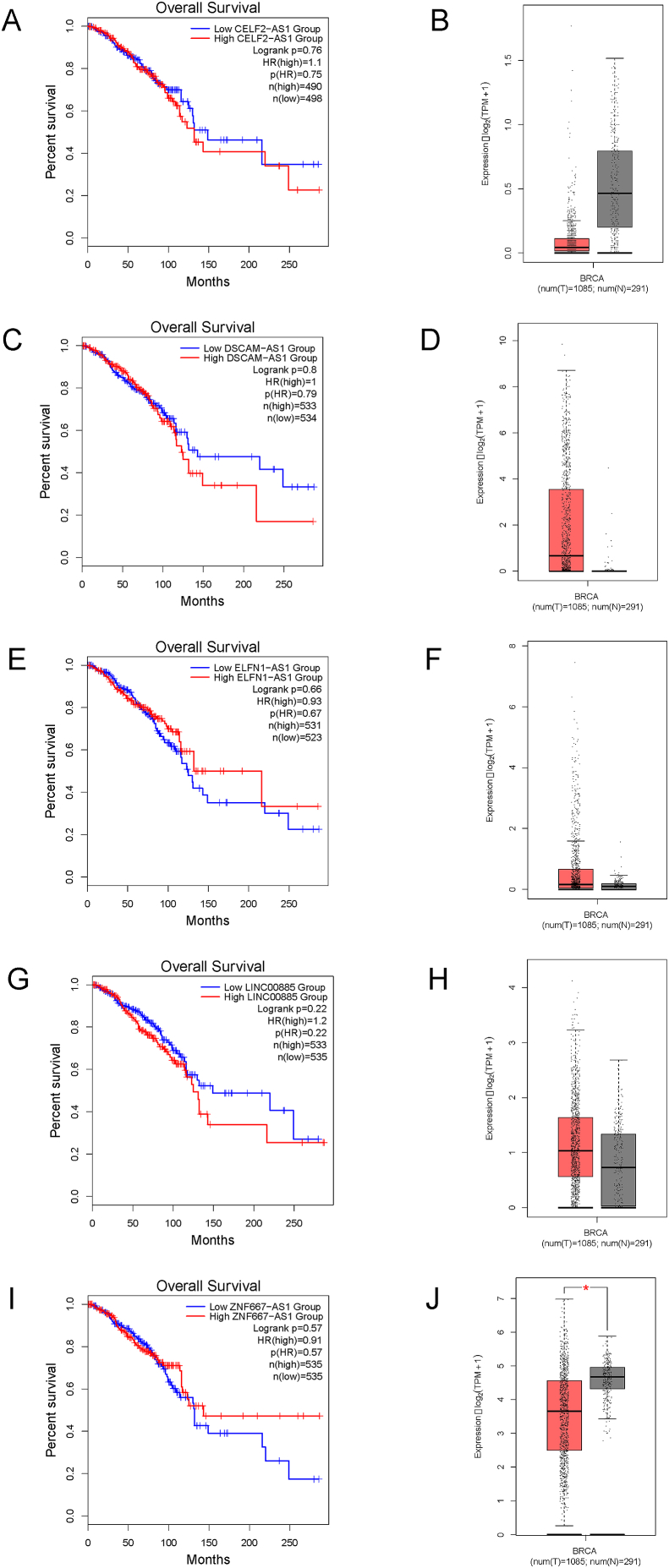
Breast cancer research has traditionally centred on genomic alterations, hormone receptor status and changes in cancer-related proteins to provide new avenues for targeted therapies. Due to advances in next generation sequencing technologies, there has been the emergence of long, non-coding RNAs (lncRNAs) as regulators of normal cellular events, with links to various disease states, including breast cancer. Here we describe our bioinformatic analyses of a previously published RNA sequencing (RNA-seq) dataset to identify lncRNAs with altered expression levels in a subset of breast cancer cell lines. Using a previously published RNA-seq dataset of 675 cancer cell lines, a subset of 18 cell lines was selected for our analyses that included 16 breast cancer lines, one ductal carcinoma line and one normal-like breast epithelial cell line. Principal component analysis demonstrated correlation with well-established categorisation methods of breast cancer (i.e. luminal A/B, HER2 enriched and basal-like A/B). Through detailed comparison of differentially expressed lncRNAs in each breast cancer sub-type with normal-like breast epithelial cells, we identified 15 lncRNAs with consistently altered expression, including three uncharacterised lncRNAs. Utilising data from The Cancer Genome Atlas (TCGA) and The Genotype Tissue Expression (GETx) project via Gene Expression Profiling Interactive Analysis (GEPIA2), we assessed clinical relevance of several identified lncRNAs with invasive breast cancer. Lastly, we determined the relative expression level of six lncRNAs across a spectrum of breast cancer cell lines to experimentally confirm the findings of our bioinformatic analyses. Overall, we show that the use of existing RNA-seq datasets, if re-analysed with modern bioinformatic tools, can provide a valuable resource to identify lncRNAs that could have important biological roles in oncogenesis and tumour progression.
乳腺癌研究传统上聚焦于基因组改变、激素受体状态以及癌症相关蛋白的变化,以探寻靶向治疗的新途径。由于新一代测序技术的进步,长链非编码RNA(lncRNA)作为正常细胞事件的调节因子出现了,它与包括乳腺癌在内的各种疾病状态相关。在此,我们描述了对先前发表的RNA测序(RNA-seq)数据集进行的生物信息学分析,以识别在一部分乳腺癌细胞系中表达水平发生改变的lncRNA。利用先前发表的包含675个癌细胞系的RNA-seq数据集,我们选择了18个细胞系进行分析,其中包括16个乳腺癌细胞系、1个导管癌细胞系和1个类正常乳腺上皮细胞系。主成分分析表明与已确立的乳腺癌分类方法(即腔面A/B型、HER2富集型和基底样A/B型)相关。通过详细比较各乳腺癌亚型中差异表达的lncRNA与类正常乳腺上皮细胞,我们鉴定出15个表达持续改变的lncRNA,其中包括3个未表征的lncRNA。通过基因表达谱交互式分析(GEPIA2)利用来自癌症基因组图谱(TCGA)和基因型组织表达(GETx)项目的数据,我们评估了几种已鉴定的lncRNA与浸润性乳腺癌的临床相关性。最后,我们测定了6个lncRNA在一系列乳腺癌细胞系中的相对表达水平,以实验性地证实我们生物信息学分析的结果。总体而言,我们表明,如果使用现代生物信息学工具对现有RNA-seq数据集进行重新分析,可为鉴定在肿瘤发生和肿瘤进展中可能具有重要生物学作用的lncRNA提供有价值的资源。