Evolutionary and Systematic Botany Group, Institute of Plant Sciences, University of Regensburg, Regensburg, Germany.
PLoS One. 2020 Mar 24;15(3):e0226234. doi: 10.1371/journal.pone.0226234. eCollection 2020.
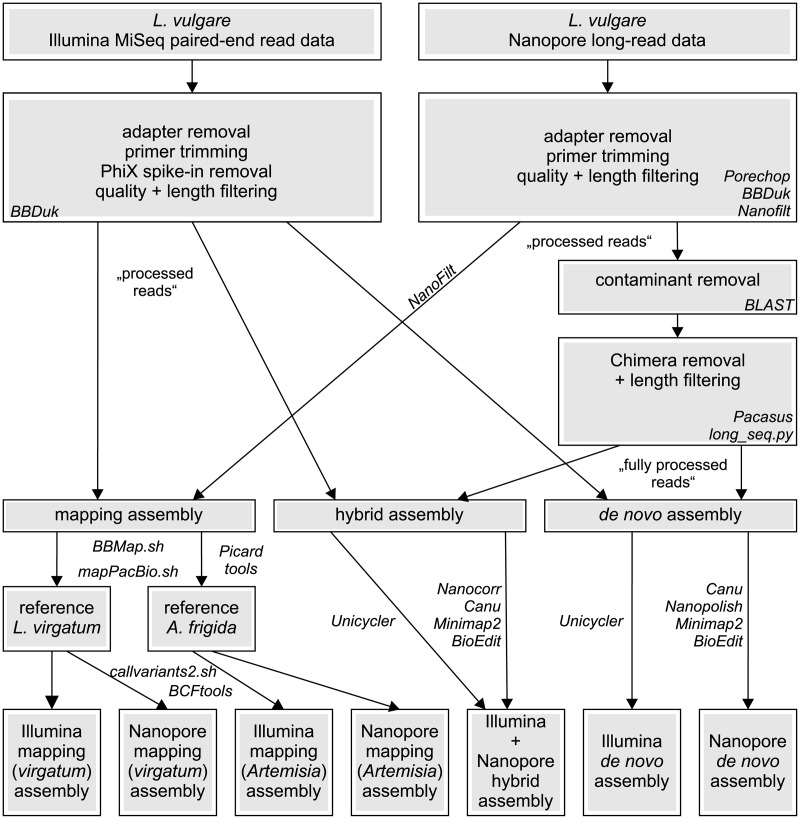
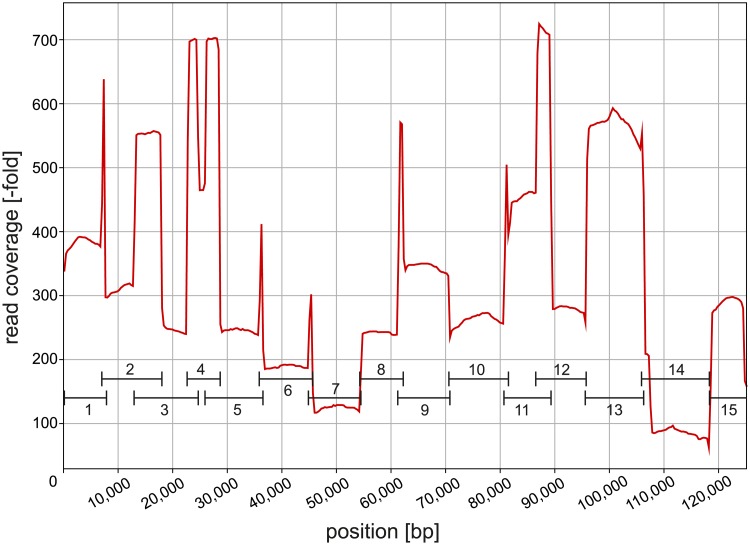
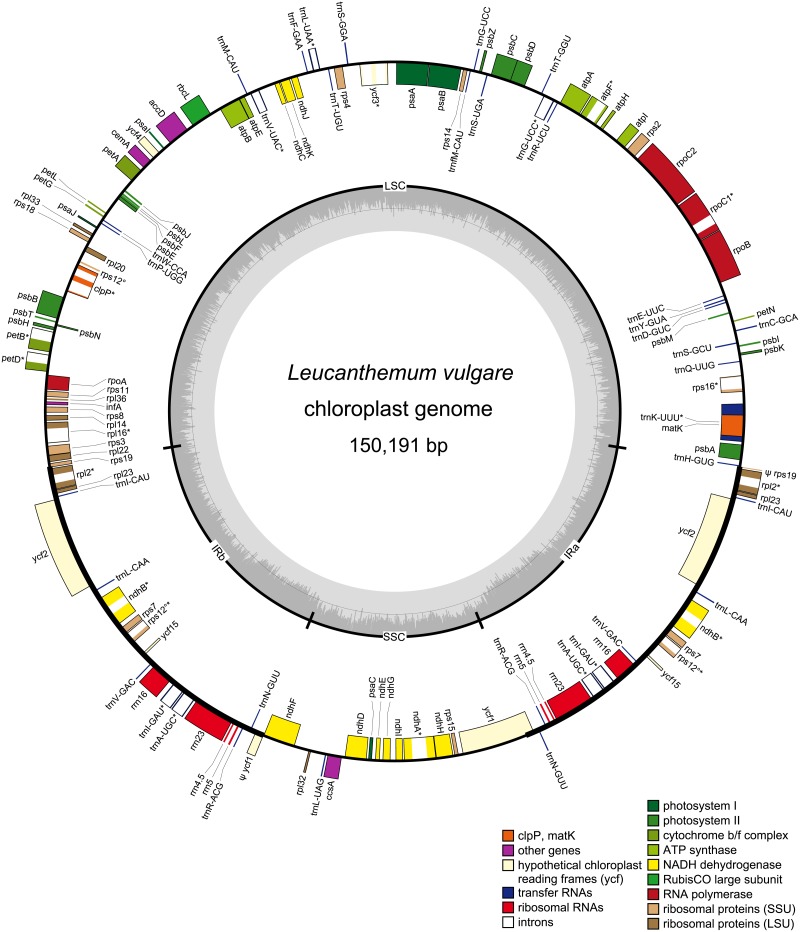
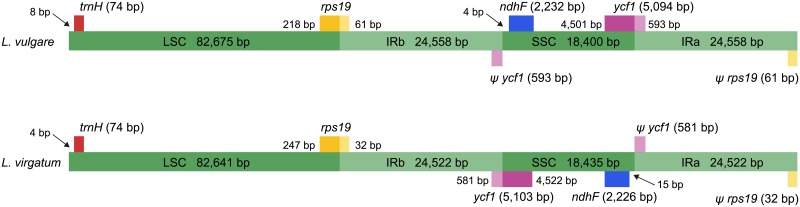
The chloroplast genome harbors plenty of valuable information for phylogenetic research. Illumina short-read data is generally used for de novo assembly of whole plastomes. PacBio or Oxford Nanopore long reads are additionally employed in hybrid approaches to enable assembly across the highly similar inverted repeats of a chloroplast genome. Unlike for PacBio, plastome assemblies based solely on Nanopore reads are rarely found, due to their high error rate and non-random error profile. However, the actual quality decline connected to their use has rarely been quantified. Furthermore, no study has employed reference-based assembly using Nanopore reads, which is common with Illumina data. Using Leucanthemum Mill. as an example, we compared the sequence quality of seven chloroplast genome assemblies of the same species, using combinations of two sequencing platforms and three analysis pipelines. In addition, we assessed the factors which might influence Nanopore assembly quality during sequence generation and bioinformatic processing. The consensus sequence derived from de novo assembly of Nanopore data had a sequence identity of 99.59% compared to Illumina short-read de novo assembly. Most of the errors detected were indels (81.5%), and a large majority of them is part of homopolymer regions. The quality of reference-based assembly is heavily dependent upon the choice of a close-enough reference. When using a reference with 0.83% sequence divergence from the studied species, mapping of Nanopore reads results in a consensus comparable to that from Nanopore de novo assembly, and of only slightly inferior quality compared to a reference-based assembly with Illumina data. For optimal de novo assembly of Nanopore data, appropriate filtering of contaminants and chimeric sequences, as well as employing moderate read coverage, is essential. Based on these results, we conclude that Nanopore long reads are a suitable alternative to Illumina short reads in plastome phylogenomics. Few errors remain in the finalized assembly, which can be easily masked in phylogenetic analyses without loss in analytical accuracy. The easily applicable and cost-effective technology might warrant more attention by researchers dealing with plant chloroplast genomes.
叶绿体基因组为系统发育研究提供了丰富的有价值信息。Illumina 短读长数据通常用于整个质体基因组的从头组装。PacBio 或 Oxford Nanopore 长读长数据还被用于混合方法,以实现叶绿体基因组高度相似的反向重复区的组装。与 PacBio 不同,由于其错误率高且错误分布不规则,很少基于 Nanopore reads 进行质体基因组组装。然而,其使用相关的实际质量下降很少被量化。此外,由于与 Illumina 数据相比,很少有研究使用基于 Nanopore reads 的参考组装。以 Leucanthemum Mill. 为例,我们比较了同一物种的七个叶绿体基因组组装的序列质量,使用了两种测序平台和三种分析管道的组合。此外,我们评估了在序列生成和生物信息处理过程中可能影响 Nanopore 组装质量的因素。与 Illumina 短读长从头组装相比,从头组装 Nanopore 数据的共识序列的序列同一性为 99.59%。大多数检测到的错误是插入缺失(81.5%),其中大多数是高度同源区的一部分。基于参考的组装质量严重依赖于接近参考的选择。当使用与研究物种序列差异为 0.83%的参考时,Nanopore reads 的映射导致的共识与 Nanopore 从头组装的共识相当,并且与使用 Illumina 数据的基于参考的组装相比质量仅略差。为了实现 Nanopore 数据的最佳从头组装,必须适当过滤污染物和嵌合体序列,并采用适度的读长覆盖度。基于这些结果,我们得出结论,Nanopore 长读长是质体基因组系统发育学中替代 Illumina 短读长的合适选择。最终组装中只有少数错误,在不影响分析准确性的情况下,很容易在系统发育分析中屏蔽这些错误。这种易于应用且具有成本效益的技术可能值得更多关注,尤其对于处理植物叶绿体基因组的研究人员而言。