Department of Biostatistics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA.
Department of Pediatrics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA.
Sci Rep. 2020 Mar 25;10(1):5434. doi: 10.1038/s41598-020-62330-2.
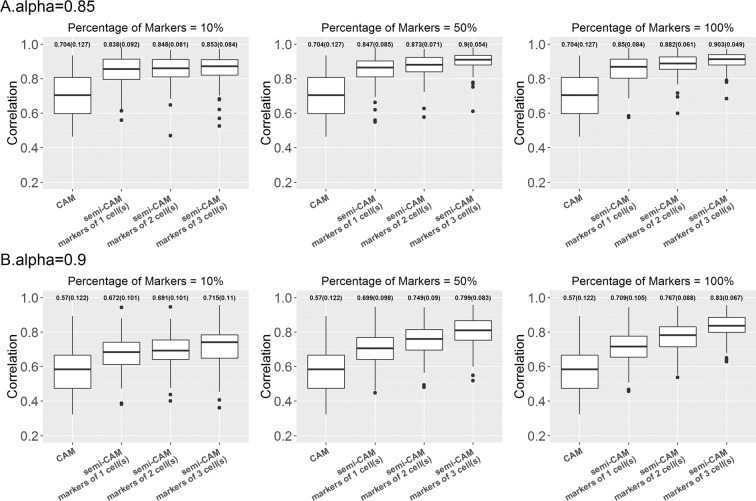
Deconvolution of bulk transcriptomics data from mixed cell populations is vital to identify the cellular mechanism of complex diseases. Existing deconvolution approaches can be divided into two major groups: supervised and unsupervised methods. Supervised deconvolution methods use cell type-specific prior information including cell proportions, reference cell type-specific gene signatures, or marker genes for each cell type, which may not be available in practice. Unsupervised methods, such as non-negative matrix factorization (NMF) and Convex Analysis of Mixtures (CAM), in contrast, completely disregard prior information and thus are not efficient for data with partial cell type-specific information. In this paper, we propose a semi-supervised deconvolution method, semi-CAM, that extends CAM by utilizing marker information from partial cell types. Analysis of simulation and two benchmark data have demonstrated that semi-CAM outperforms CAM by yielding more accurate cell proportion estimations when markers from partial/all cell types are available. In addition, when markers from all cell types are available, semi-CAM achieves better or similar accuracy compared to the supervised method using signature genes, CIBERSORT, and the marker-based supervised methods semi-NMF and DSA. Furthermore, analysis of human chlamydia-infection data with bulk expression profiles from six cell types and prior marker information of only three cell types suggests that semi-CAM achieves more accurate cell proportion estimations than CAM.
从混合细胞群体中反卷积批量转录组学数据对于识别复杂疾病的细胞机制至关重要。现有的反卷积方法可分为两大类:有监督和无监督方法。有监督反卷积方法使用细胞类型特异性的先验信息,包括细胞比例、参考细胞类型特异性基因特征或每个细胞类型的标记基因,但在实践中可能无法获得。相比之下,无监督方法(如非负矩阵分解(NMF)和混合物凸分析(CAM))完全忽略了先验信息,因此对于具有部分细胞类型特异性信息的数据效率不高。在本文中,我们提出了一种半监督反卷积方法 semi-CAM,它通过利用部分细胞类型的标记信息扩展了 CAM。通过对模拟数据和两个基准数据集的分析,我们证明了当部分/所有细胞类型的标记可用时,semi-CAM 比 CAM 能够产生更准确的细胞比例估计。此外,当所有细胞类型的标记都可用时,与使用特征基因、CIBERSORT 的有监督方法以及基于标记的有监督方法 semi-NMF 和 DSA 相比,semi-CAM 具有更好或相似的准确性。此外,对来自六个细胞类型的批量表达谱和仅三个细胞类型的先验标记信息的人类衣原体感染数据进行分析表明,semi-CAM 比 CAM 能够实现更准确的细胞比例估计。