Lunenfeld-Tanenbaum Research Institute, Sinai Health System, Toronto, Ontario, Canada.
Centre for Healthcare Innovation, Rady Faculty of Health Science, University of Manitoba, Winnipeg, Manitoba, Canada.
Genet Epidemiol. 2020 Jun;44(4):368-381. doi: 10.1002/gepi.22291. Epub 2020 Apr 1.
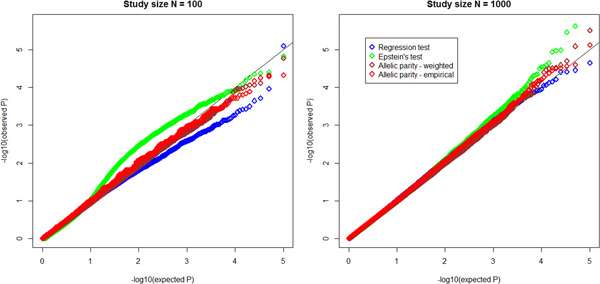
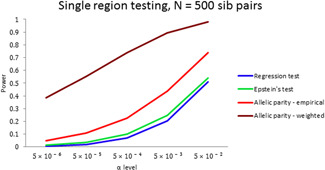
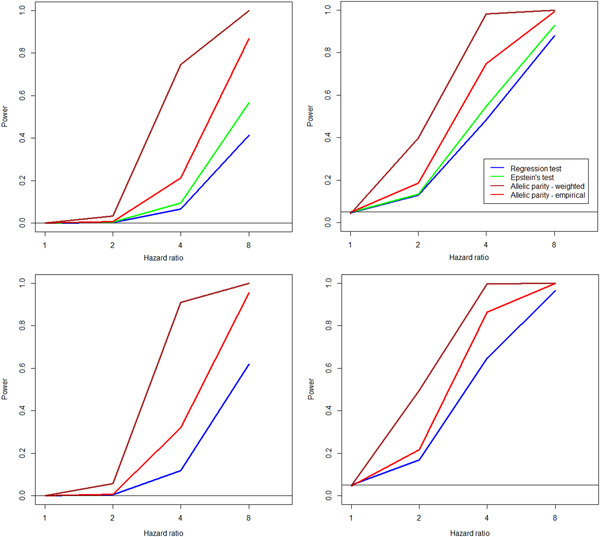
Next generation sequencing technologies have made it possible to investigate the role of rare variants (RVs) in disease etiology. Because RVs associated with disease susceptibility tend to be enriched in families with affected individuals, study designs based on affected sib pairs (ASP) can be more powerful than case-control studies. We construct tests of RV-set association in ASPs for single genomic regions as well as for multiple regions. Single-region tests can efficiently detect a gene region harboring susceptibility variants, while multiple-region extensions are meant to capture signals dispersed across a biological pathway, potentially as a result of locus heterogeneity. Within ascertained ASPs, the test statistics contrast the frequencies of duplicate rare alleles (usually appearing on a shared haplotype) against frequencies of a single rare allele copy (appearing on a nonshared haplotype); we call these allelic parity tests. Incorporation of minor allele frequency estimates from reference populations can markedly improve test efficiency. Under various genetic penetrance models, application of the tests in simulated ASP data sets demonstrates good type I error properties as well as power gains over approaches that regress ASP rare allele counts on sharing state, especially in small samples. We discuss robustness of the allelic parity methods to the presence of genetic linkage, misspecification of reference population allele frequencies, sequencing error and de novo mutations, and population stratification. As proof of principle, we apply single- and multiple-region tests in a motivating study data set consisting of whole exome sequencing of sisters ascertained with early onset breast cancer.
下一代测序技术使得研究罕见变异(RVs)在疾病病因学中的作用成为可能。由于与疾病易感性相关的 RVs 往往在受影响个体的家族中富集,因此基于受影响同胞对(ASP)的研究设计比病例对照研究更有效。我们构建了用于 ASP 中单基因组区域和多个区域的 RV 集关联的测试。单区域测试可以有效地检测到携带易感变异的基因区域,而多区域扩展旨在捕获分散在生物途径中的信号,可能是由于基因座异质性。在确定的 ASP 中,测试统计数据对比了重复稀有等位基因(通常出现在共享单倍型上)的频率与单个稀有等位基因副本(出现在非共享单倍型上)的频率;我们称这些等位基因奇偶校验测试。从参考群体中纳入小等位基因频率估计值可以显著提高测试效率。在各种遗传穿透性模型下,将测试应用于模拟 ASP 数据集表明,与回归 ASP 稀有等位基因计数与共享状态的方法相比,具有良好的Ⅰ型错误属性和功效增益,尤其是在小样本中。我们讨论了等位基因奇偶校验方法对遗传连锁、参考群体等位基因频率的不正确指定、测序错误和新生突变以及群体分层的稳健性。作为原理验证,我们将单区域和多区域测试应用于由姐妹全外显子组测序组成的有启发性的研究数据集,这些姐妹的发病年龄较早。