Center for Precision Disease Modeling, Department of Medicine, University of Maryland School of Medicine, Baltimore, Maryland.
Division of Endocrinology, Diabetes and Nutrition, Department of Medicine, University of Maryland School of Medicine, Baltimore, Maryland.
J Am Soc Nephrol. 2020 May;31(5):1024-1034. doi: 10.1681/ASN.2019060591. Epub 2020 Apr 1.
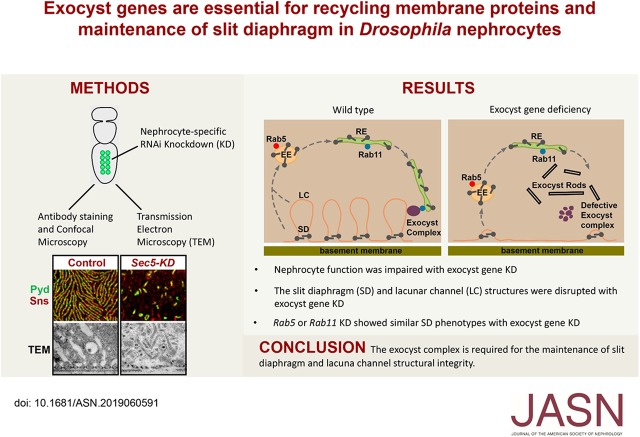
Studies have linked mutations in genes encoding the eight-protein exocyst protein complex to kidney disease, but the underlying mechanism is unclear. Because nephrocytes share molecular and structural features with mammalian podocytes, they provide an efficient model for studying this issue.
We silenced genes encoding exocyst complex proteins specifically in nephrocytes and studied the effects on protein reabsorption by lacuna channels and filtration by the slit diaphragm. We performed nephrocyte functional assays, carried out super-resolution confocal microscopy of slit diaphragm proteins, and used transmission electron microscopy to analyze ultrastructural changes. We also examined the colocalization of slit diaphragm proteins with exocyst protein Sec15 and with endocytosis and recycling regulators Rab5, Rab7, and Rab11.
Silencing exocyst genes in nephrocytes led to profound changes in structure and function. Abolition of cellular accumulation of hemolymph proteins with dramatically reduced lacuna channel membrane invaginations offered a strong indication of reabsorption defects. Moreover, the slit diaphragm's highly organized surface structure-essential for filtration-was disrupted, and key proteins were mislocalized. Ultrastructural analysis revealed that exocyst gene silencing led to the striking appearance of novel electron-dense structures that we named "exocyst rods," which likely represent accumulated membrane proteins following defective exocytosis or recycling. The slit diaphragm proteins partially colocalized with Sec15, Rab5, and Rab11.
Our findings suggest that the slit diaphragm of nephrocytes requires balanced endocytosis and recycling to maintain its structural integrity and that impairment of the exocyst complex leads to disruption of the slit diaphragm and nephrocyte malfunction. This model may help identify therapeutic targets for treating kidney diseases featuring molecular defects in vesicle endocytosis, exocytosis, and recycling.
研究将编码八蛋白外泌体蛋白复合物的基因突变与肾脏疾病联系起来,但潜在机制尚不清楚。由于肾单位与哺乳动物足细胞具有分子和结构特征,因此它们为研究这一问题提供了有效的模型。
我们特异性地在肾单位中沉默编码外泌体复合物蛋白的基因,并研究其对腔道蛋白重吸收和裂隙隔膜过滤的影响。我们进行了肾单位功能测定,对裂隙隔膜蛋白进行超分辨率共聚焦显微镜观察,并使用透射电子显微镜分析超微结构变化。我们还检查了裂隙隔膜蛋白与外泌体蛋白 Sec15以及内吞和再循环调节因子 Rab5、Rab7 和 Rab11 的共定位。
沉默肾单位中的外泌体基因导致结构和功能发生深刻变化。消除血液蛋白的细胞积累,并显著减少腔道蛋白膜内陷,强烈提示重吸收缺陷。此外,裂隙隔膜的高度有序表面结构(对过滤至关重要)被破坏,关键蛋白发生定位错误。超微结构分析显示,外泌体基因沉默导致出现了新型电子致密结构,我们称之为“外泌体棒”,这可能代表了内吐或再循环缺陷后积累的膜蛋白。裂隙隔膜蛋白与 Sec15、Rab5 和 Rab11 部分共定位。
我们的研究结果表明,肾单位的裂隙隔膜需要平衡的内吞和再循环来维持其结构完整性,而外泌体复合物的损伤会导致裂隙隔膜和肾单位功能障碍。该模型可能有助于确定治疗以囊泡内吞、外吐和再循环分子缺陷为特征的肾脏疾病的治疗靶点。