Department of Pharmaceutical Chemistry, R.C. Patel Institute of Pharmaceutical Education and Research, Shirpur, Dist-Dhule 425405, Maharashtra, India.
Department of Chemical Technology, Dr. Babasaheb Ambedkar Marathwada University, Aurangabad, 431004 Maharashtra, India.
Molecules. 2020 Apr 1;25(7):1622. doi: 10.3390/molecules25071622.
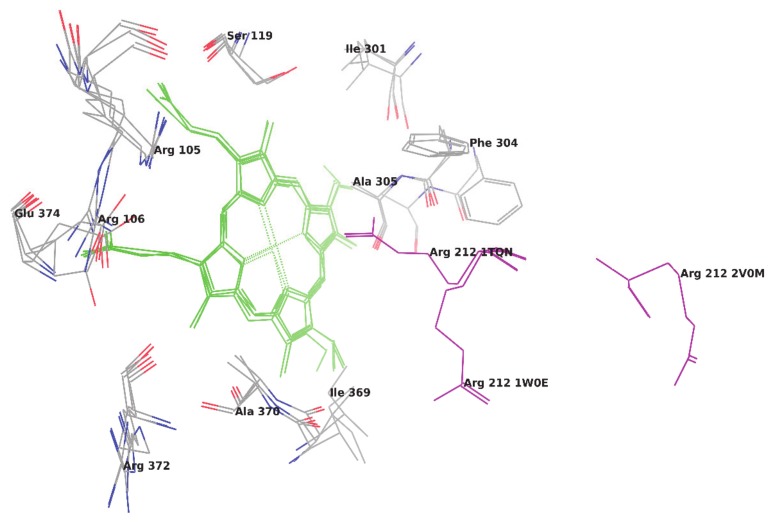
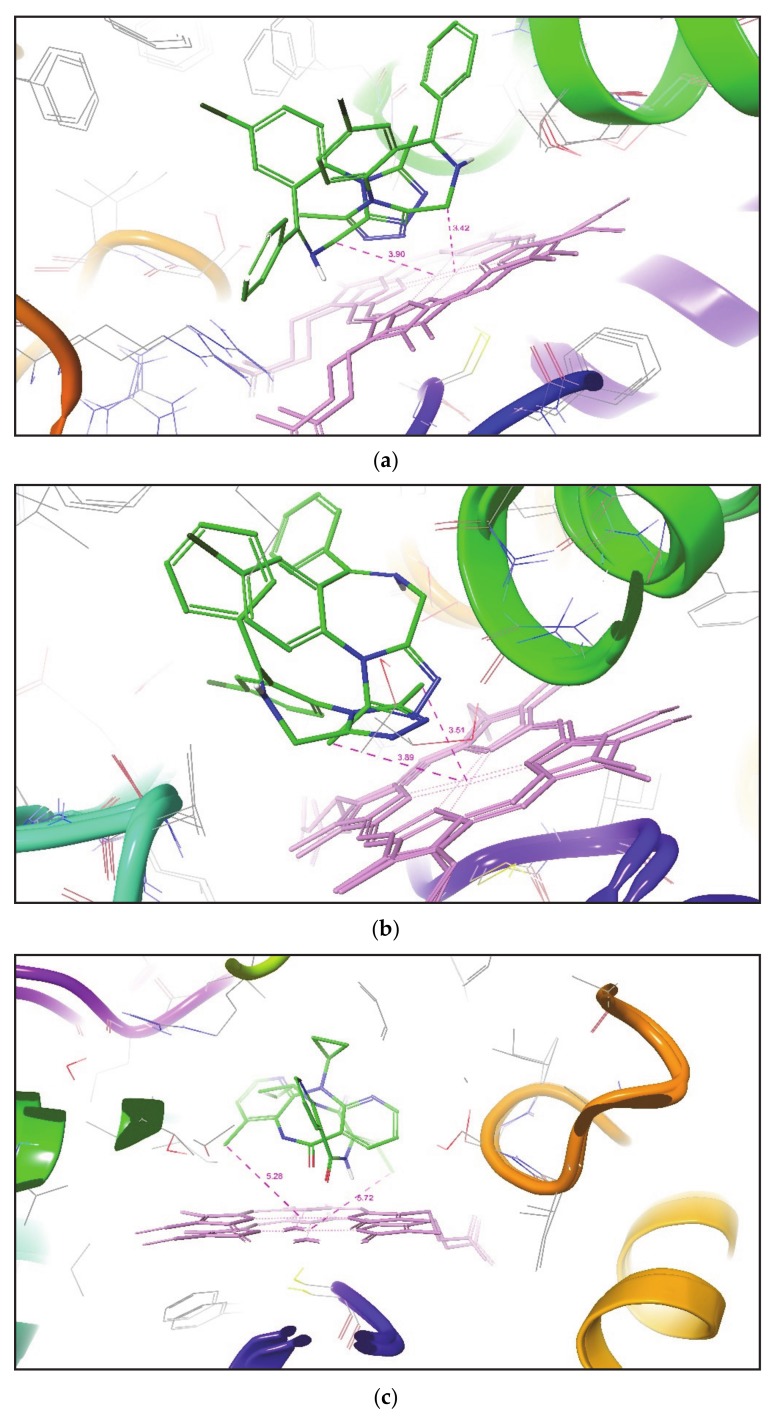
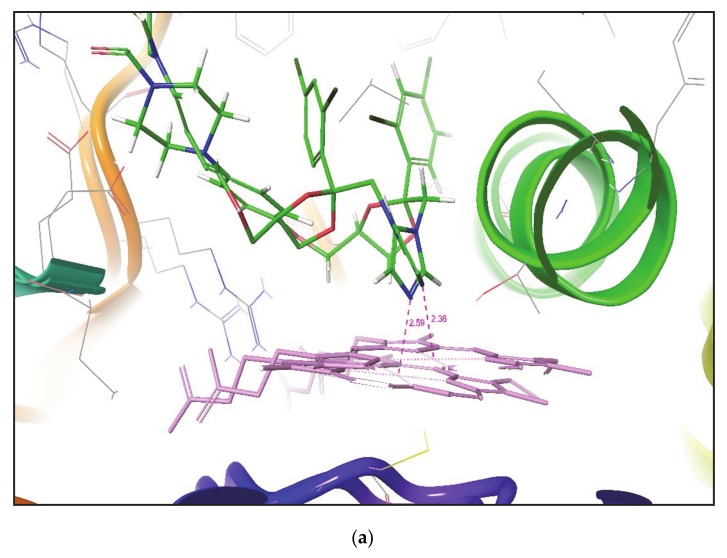
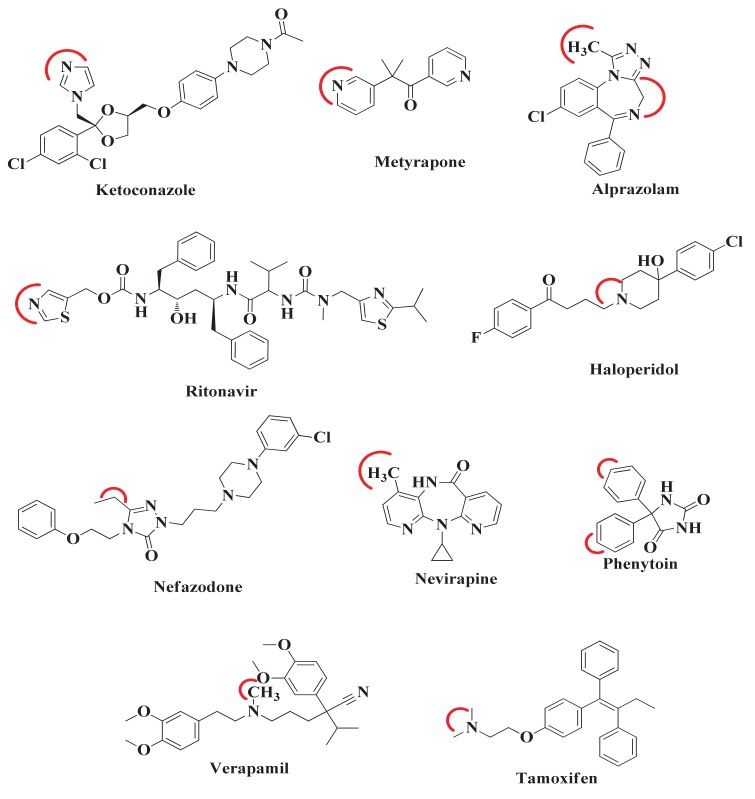
Metabolism is one of the prime reasons where most of drugs fail to accomplish their clinical trials. The enzyme CYP3A4, which belongs to the superfamily of cytochrome P450 enzymes (CYP), helps in the metabolism of a large number of drugs in the body. The enzyme CYP3A4 catalyzes oxidative chemical processes and shows a very broad range of ligand specificity. The understanding of the compound's structure where oxidation would take place is crucial for the successful modification of molecules to avoid unwanted metabolism and to increase its bioavailability. For this reason, it is required to know the site of metabolism (SOM) of the compounds, where compounds undergo enzymatic oxidation. It can be identified by predicting the accessibility of the substrate's atom toward oxygenated Fe atom of heme in a CYP protein. The CYP3A4 enzyme is highly flexible and can take significantly different conformations depending on the ligand with which it is being bound. To predict the accessibility of substrate atoms to the heme iron, conventional protein-rigid docking methods failed due to the high flexibility of the CYP3A4 protein. Herein, we demonstrated and compared the ability of the Glide extra precision (XP) and Induced Fit docking (IFD) tool of Schrodinger software suite to reproduce the binding mode of co-crystallized ligands into six X-ray crystallographic structures. We extend our studies toward the prediction of SOM for compounds whose experimental SOM is reported but the ligand-enzyme complex crystal structure is not available in the Protein Data Bank (PDB). The quality and accuracy of Glide XP and IFD was determined by calculating RMSD of docked ligands over the corresponding co-crystallized bound ligand and by measuring the distance between the SOM of the ligand and Fe atom of heme. It was observed that IFD reproduces the exact binding mode of available co-crystallized structures and correctly predicted the SOM of experimentally reported compounds. Our approach using IFD with multiple conformer structures of CYP3A4 will be one of the effective methods for SOM prediction.
代谢是大多数药物临床试验失败的主要原因之一。CYP3A4 酶属于细胞色素 P450 酶超家族(CYP),有助于体内许多药物的代谢。CYP3A4 酶催化氧化化学过程,表现出非常广泛的配体特异性。了解发生氧化的化合物结构对于成功修饰分子以避免不必要的代谢并提高其生物利用度至关重要。出于这个原因,需要知道化合物的代谢部位(SOM),即化合物发生酶氧化的部位。可以通过预测底物原子对 CYP 蛋白中血红素含氧 Fe 原子的可及性来识别。CYP3A4 酶具有高度的灵活性,并且可以根据与其结合的配体而呈现出显著不同的构象。为了预测底物原子对血红素铁的可及性,传统的蛋白质刚性对接方法由于 CYP3A4 蛋白质的高灵活性而失败。在这里,我们展示并比较了 Schrödinger 软件套件中的 Glide 超高精度(XP)和诱导契合对接(IFD)工具复制共结晶配体结合模式的能力到六个 X 射线晶体结构中。我们将研究扩展到预测 SOM 的化合物,这些化合物的实验 SOM 已报告,但配体-酶复合物晶体结构在蛋白质数据银行(PDB)中不可用。通过计算对接配体相对于相应共结晶结合配体的 RMSD 并测量配体的 SOM 与血红素铁原子之间的距离,来确定 Glide XP 和 IFD 的质量和准确性。观察到 IFD 重现了可用共结晶结构的精确结合模式,并正确预测了实验报告化合物的 SOM。我们使用 IFD 结合 CYP3A4 的多种构象结构的方法将成为 SOM 预测的有效方法之一。