Institut des Hautes Études Scientifiques, Bures-sur-Yvette, France.
Mathematics Department, University of California at Los Angeles, Los Angeles, California.
J Comput Biol. 2020 Oct;27(10):1495-1508. doi: 10.1089/cmb.2020.0120. Epub 2020 Apr 3.
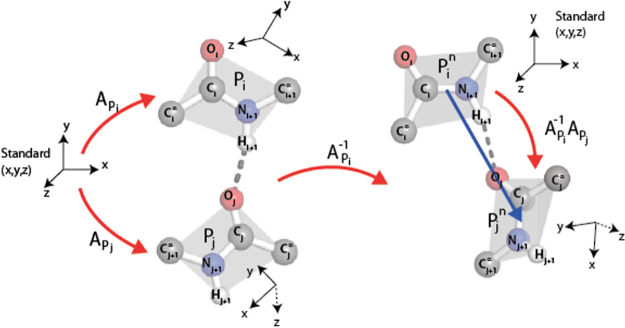
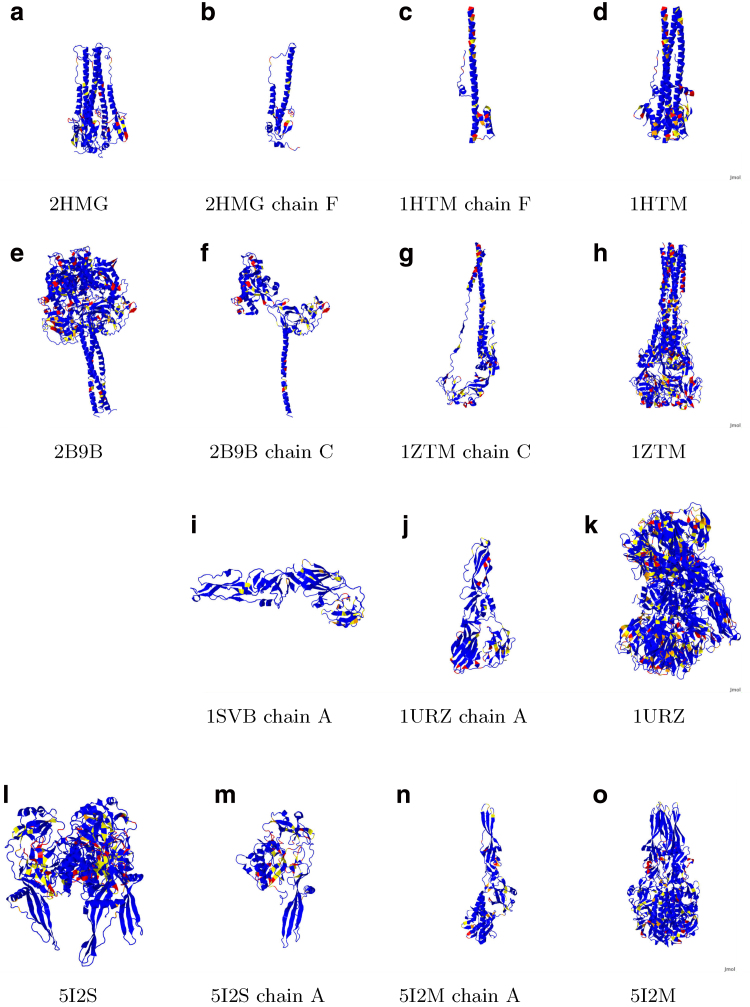
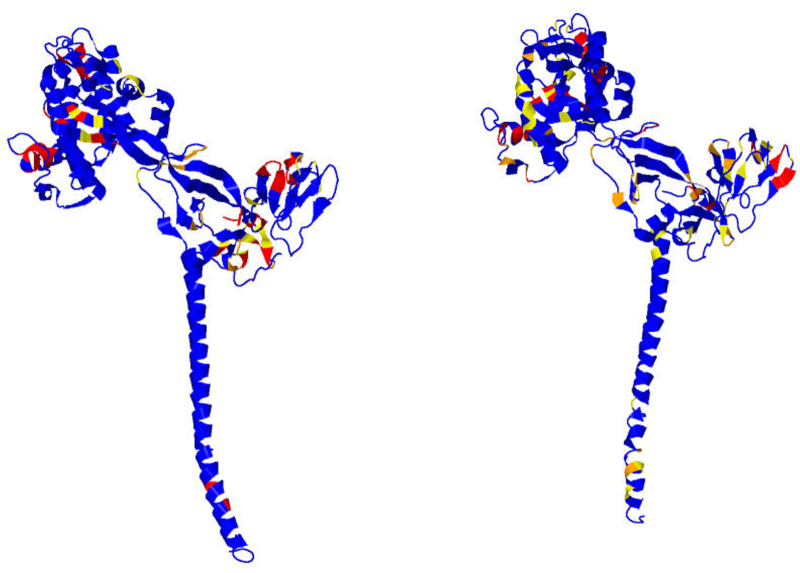
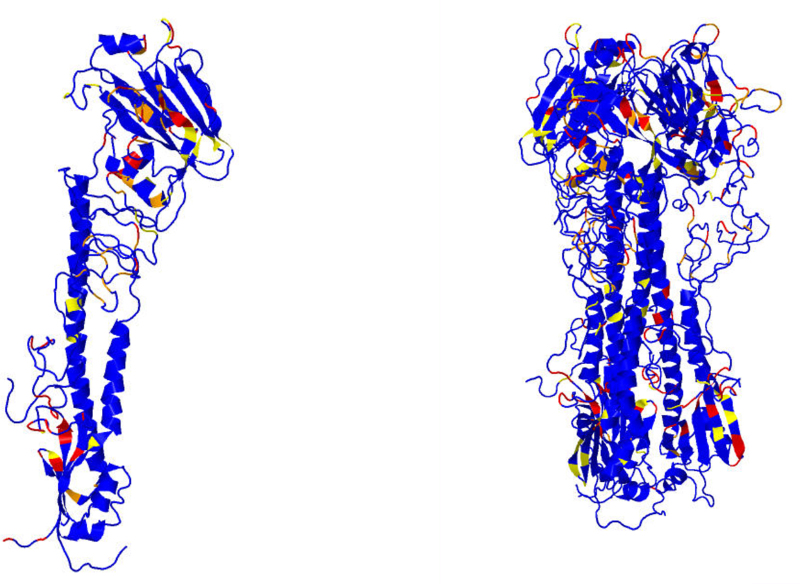
Earlier analysis of the Protein Data Bank derived the distribution of rotations from the plane of a protein hydrogen bond donor peptide group to the plane of its acceptor peptide group. The quasi Boltzmann formalism of Pohl-Finkelstein is employed to estimate free energies of protein elements with these hydrogen bonds, pinpointing residues with a high propensity for conformational change. This is applied to viral glycoproteins as well as capsids, where the 90th+ percentiles of free energies determine residues that correlate well with viral fusion peptides and other functional domains in known cases and thus provide a novel method for predicting these sites of importance as antiviral drug or vaccine targets in general. The method is implemented at https://bion-server.au.dk/hbonds/ from an uploaded Protein Data Bank file.
先前对蛋白质数据库的分析得出了蛋白质氢键供体肽基团平面到受体肽基团平面的旋转分布。采用 Pohl-Finkelstein 的准玻尔兹曼形式主义来估计具有这些氢键的蛋白质元素的自由能,精确定位具有高构象变化倾向的残基。该方法适用于病毒糖蛋白和衣壳,其中自由能的 90 百分位数确定与已知情况下的病毒融合肽和其他功能域相关的残基,从而为预测这些重要的抗病毒药物或疫苗靶点提供了一种新方法。该方法可从上传的蛋白质数据库文件在 https://bion-server.au.dk/hbonds/ 上执行。