South African Medical Research Council (SAMRC): Respiratory and Meningeal Pathogens Research Unit, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa; Department of Science and Technology/National Research Foundation (DST/NRF): Vaccine Preventable Diseases, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa.
Wits Reproductive Health and HIV Institute, School of Clinical Medicine, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa.
Lancet Infect Dis. 2020 Jul;20(7):851-863. doi: 10.1016/S1473-3099(20)30001-3. Epub 2020 Apr 3.
A monovalent, parenteral, subunit rotavirus vaccine was well tolerated and immunogenic in adults in the USA and in toddlers and infants in South Africa, but elicited poor responses against heterotypic rotavirus strains. We aimed to evaluate safety and immunogenicity of a trivalent vaccine formulation (P2-VP8-P[4],[6],[8]).
A double-blind, randomised, placebo-controlled, dose-escalation, phase 1/2 study was done at three South African research sites. Healthy adults (aged 18-45 years), toddlers (aged 2-3 years), and infants (aged 6-8 weeks, ≥37 weeks' gestation, and without previous receipt of rotavirus vaccination), all without HIV infection, were eligible for enrolment. In the dose-escalation phase, adults and toddlers were randomly assigned in blocks (block size of five) to receive 30 μg or 90 μg of vaccine, or placebo, and infants were randomly assigned in blocks (block size of four) to receive 15 μg, 30 μg, or 90 μg of vaccine, or placebo. In the expanded phase, infants were randomly assigned in a 1:1:1:1 ratio to receive 15 μg, 30 μg, or 90 μg of vaccine, or placebo, in block sizes of four. Participants, parents of participants, and clinical, data, and laboratory staff were masked to treatment assignment. Adults received an intramuscular injection of vaccine or placebo in the deltoid muscle on the day of randomisation (day 0), day 28, and day 56; toddlers received a single injection of vaccine or placebo in the anterolateral thigh on day 0. Infants in both phases received an injection of vaccine or placebo in the anterolateral thigh on days 0, 28, and 56, at approximately 6, 10, and 14 weeks of age. Primary safety endpoints were local and systemic reactions (grade 2 or worse) within 7 days and adverse events and serious adverse events within 28 days after each injection in all participants who received at least one injection. Primary immunogenicity endpoints were analysed in infants in either phase who received all planned injections, had blood samples analysed at the relevant timepoints, and presented no major protocol violations considered to have an effect on the immunogenicity results of the study, and included serum anti-P2-VP8 IgA, IgG, and neutralising antibody geometric mean titres and responses measured 4 weeks after the final injection in vaccine compared with placebo groups. This trial is registered with ClinicalTrials.gov, NCT02646891.
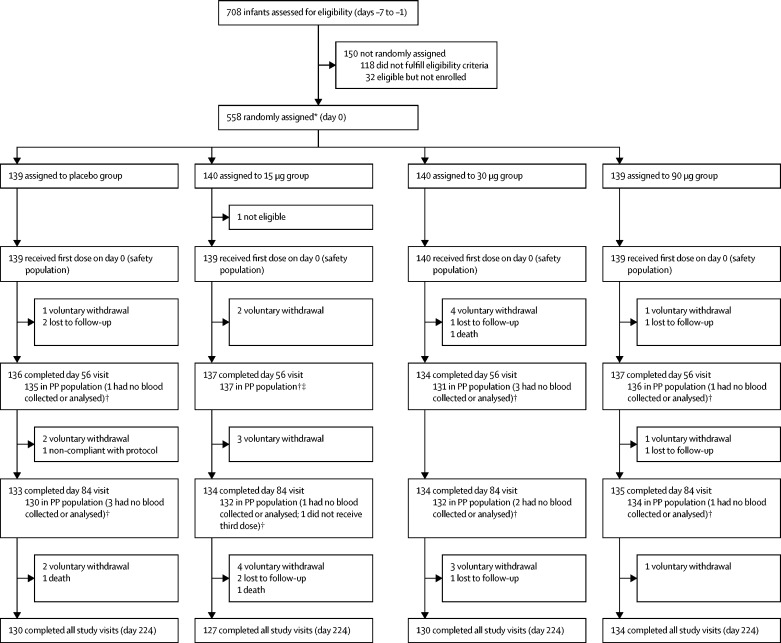
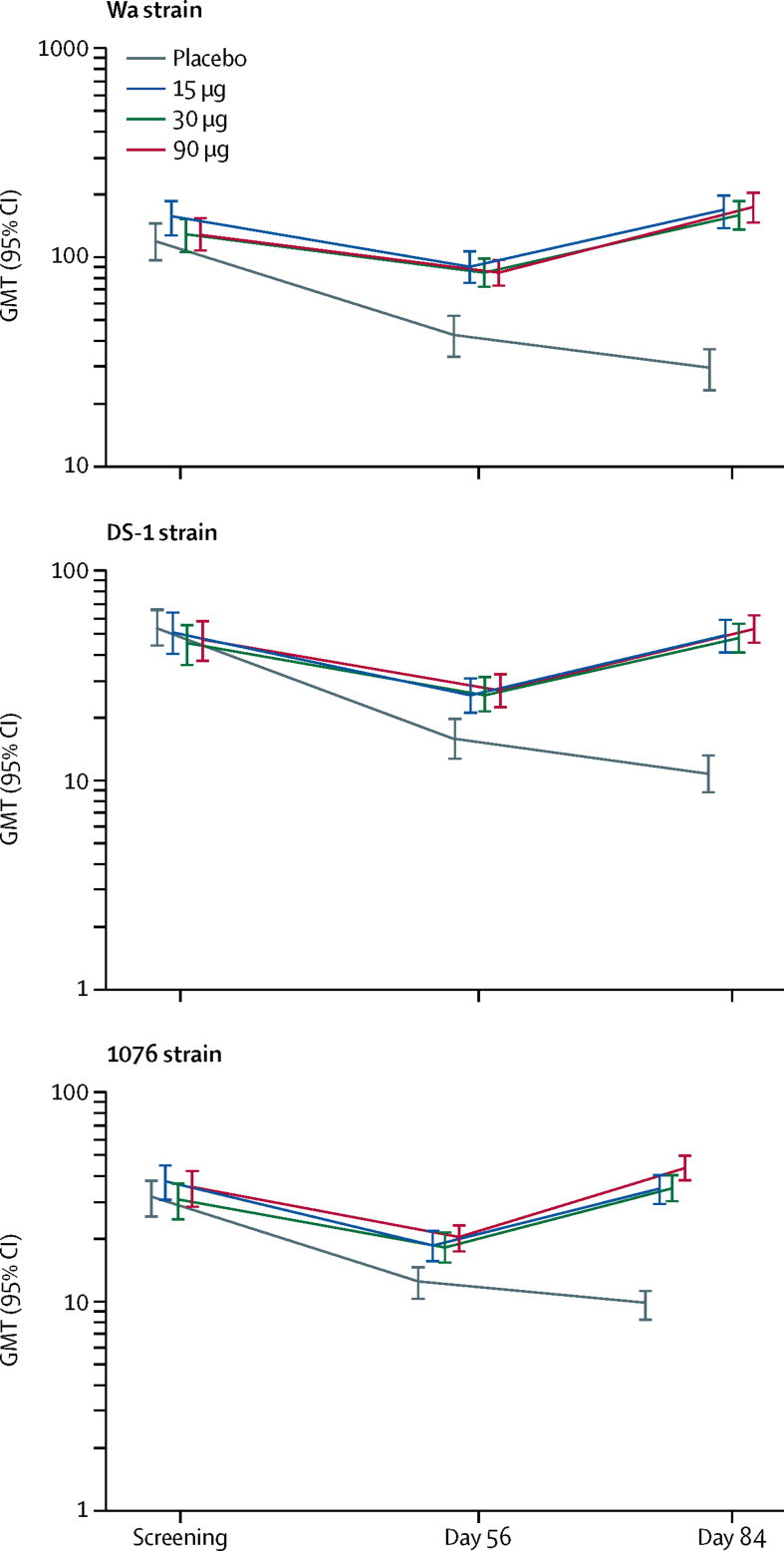
Between Feb 15, 2016, and Dec 22, 2017, 30 adults (12 each in the 30 μg and 90 μg groups and six in the placebo group), 30 toddlers (12 each in the 30 μg and 90 μg groups and six in the placebo group), and 557 infants (139 in the 15 μg group, 140 in the 30 μg group, 139 in the 90 μg group, and 139 in the placebo group) were randomly assigned, received at least one dose, and were assessed for safety. There were no significant differences in local or systemic adverse events, or unsolicited adverse events, between vaccine and placebo groups. There were no serious adverse events within 28 days of injection in adults, whereas one serious adverse event occurred in a toddler (febrile convulsion in the 30 μg group) and 23 serious adverse events (four in placebo, ten in 15 μg, four in 30 μg, and five in 90 μg groups) occurred among 20 infants, most commonly respiratory tract infections. One death occurred in an infant within 28 days of injection due to pneumococcal meningitis. In 528 infants (130 in placebo, 132 in 15 μg, 132 in 30 μg, and 134 in 90 μg groups), adjusted anti-P2-VP8 IgG seroresponses (≥4-fold increase from baseline) to P[4], P[6], and P[8] antigens were significantly higher in the 15 μg, 30 μg, and 90 μg groups (99-100%) than in the placebo group (10-29%; p<0·0001). Although significantly higher than in placebo recipients (9-10%), anti-P2-VP8 IgA seroresponses (≥4-fold increase from baseline) to each individual antigen were modest (20-34%) across the 15 μg, 30 μg, and 90 μg groups. Adjusted neutralising antibody seroresponses in infants (≥2·7-fold increase from baseline) to DS-1 (P[4]), 1076 (P[6]), and Wa (P[8]) were higher in vaccine recipients than in placebo recipients: p<0·0001 for all comparisons.
The trivalent P2-VP8 vaccine was well tolerated, with promising anti-P2-VP8 IgG and neutralising antibody responses across the three vaccine P types. Our findings support advancing the vaccine to efficacy testing.
Bill & Melinda Gates Foundation.
在美国,一种单价、肠胃外、亚单位轮状病毒疫苗在成年人和南非的幼儿及婴儿中具有良好的耐受性和免疫原性,但对异型轮状病毒株的反应较差。我们旨在评估一种三价疫苗配方(P2-VP8-P[4]、[6]、[8])的安全性和免疫原性。
在南非的三个研究地点进行了一项双盲、随机、安慰剂对照、剂量递增、1/2 期的研究。健康的成年人(年龄 18-45 岁)、幼儿(年龄 2-3 岁)和婴儿(年龄 6-8 周,≥37 周妊娠,且以前未接种过轮状病毒疫苗),均无 HIV 感染,有资格入组。在剂量递增阶段,成年人和幼儿以区块(区块大小为五)随机分配接受 30μg 或 90μg 疫苗或安慰剂,婴儿以区块(区块大小为四)随机分配接受 15μg、30μg 或 90μg 疫苗或安慰剂。在扩展阶段,婴儿以 1:1:1:1 的比例随机分配接受 15μg、30μg 或 90μg 疫苗或安慰剂,区块大小为四。参与者、参与者的父母、临床、数据和实验室工作人员对治疗分配进行了屏蔽。成年人在随机分组(第 0 天)、第 28 天和第 56 天在三角肌肌肉中接受一次肌肉注射疫苗或安慰剂;幼儿在第 0 天接受一次大腿前外侧的单剂量疫苗或安慰剂注射。在两个阶段中,婴儿在第 0、28 和 56 天在大腿前外侧接受一次注射疫苗或安慰剂,大约在 6、10 和 14 周龄时。主要安全性终点是所有接受至少一次注射的参与者在 7 天内出现的局部和全身反应(2 级或更高级别)和 28 天内的不良事件和严重不良事件。主要免疫原性终点是分析两个阶段中接受了所有计划注射、在相关时间点有血液样本分析且未出现被认为对研究免疫原性结果有影响的主要方案违规的婴儿,包括血清抗 P2-VP8 IgA、IgG 和中和抗体几何平均滴度和与安慰剂组相比,最后一次注射后 4 周的反应。这项试验在 ClinicalTrials.gov 上注册,编号为 NCT02646891。
在 2016 年 2 月 15 日至 2017 年 12 月 22 日期间,30 名成年人(每组 12 名,30μg 和 90μg 组各 6 名,安慰剂组 6 名)、30 名幼儿(每组 12 名,30μg 和 90μg 组各 6 名,安慰剂组 6 名)和 557 名婴儿(15μg 组 139 名,30μg 组 140 名,90μg 组 139 名,安慰剂组 139 名)被随机分配,接受了至少一剂疫苗,并进行了安全性评估。疫苗组和安慰剂组之间在局部或全身不良事件或未报告的不良事件方面无显著差异。在成年人中,注射后 28 天内无严重不良事件,而在一名幼儿(30μg 组发热性惊厥)和 20 名婴儿(4 名在安慰剂组,10 名在 15μg 组,4 名在 30μg 组,5 名在 90μg 组)中发生了 23 例严重不良事件(4 例在安慰剂组,10 例在 15μg 组,4 例在 30μg 组,5 例在 90μg 组),最常见的是呼吸道感染。一名婴儿在注射后 28 天内因肺炎球菌脑膜炎死亡。在 528 名婴儿(安慰剂组 130 名,15μg 组 132 名,30μg 组 132 名,90μg 组 134 名)中,15μg、30μg 和 90μg 组针对 P[4]、P[6]和 P[8]抗原的调整后抗 P2-VP8 IgG 血清应答(与基线相比增加≥4 倍)显著高于安慰剂组(10-29%;p<0·0001)。尽管与安慰剂组(9-10%)相比,抗 P2-VP8 IgA 血清应答(与基线相比增加≥4 倍)在 15μg、30μg 和 90μg 组中较低(20-34%),但在各个抗原中都有一定程度的提高。与安慰剂组相比,婴儿(与基线相比增加≥2.7 倍)针对 DS-1(P[4])、1076(P[6])和 Wa(P[8])的调整后中和抗体血清应答更高:所有比较均为 p<0·0001。
三价 P2-VP8 疫苗具有良好的耐受性,在三种疫苗 P 型中均具有良好的抗 P2-VP8 IgG 和中和抗体反应。我们的研究结果支持将疫苗推进到疗效测试阶段。
比尔和梅琳达盖茨基金会。