Centre for Theoretical Chemistry and Physics, School of Natural and Computational Sciences, Massey University Auckland, Auckland, New Zealand.
Bioinformatics Institute, Agency for Science, Technology and Research, Singapore.
Mol Biol Evol. 2020 Sep 1;37(9):2711-2726. doi: 10.1093/molbev/msaa100.
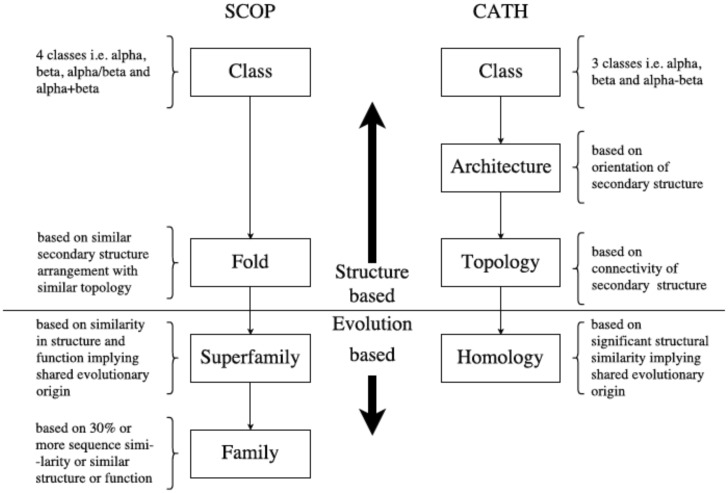
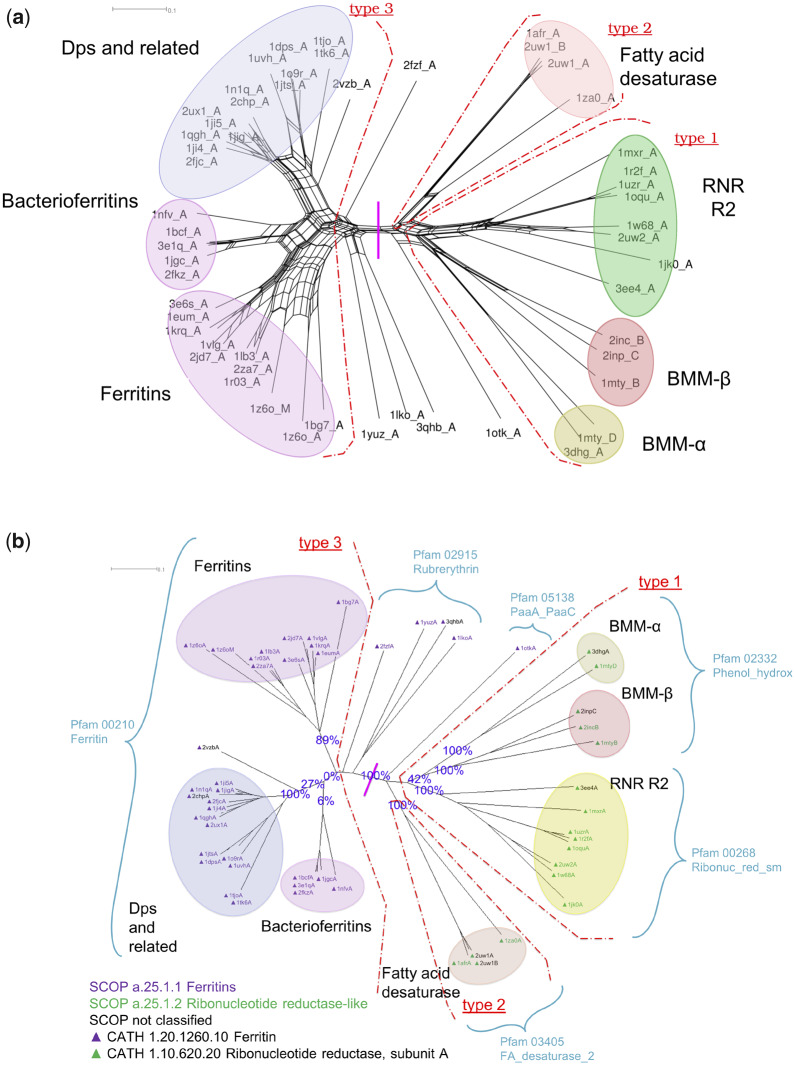
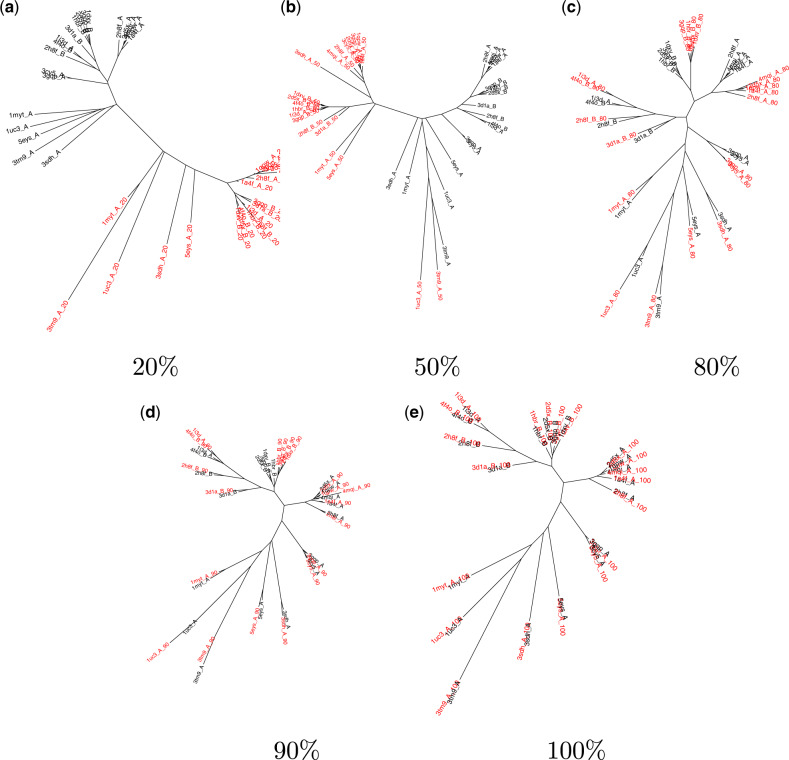
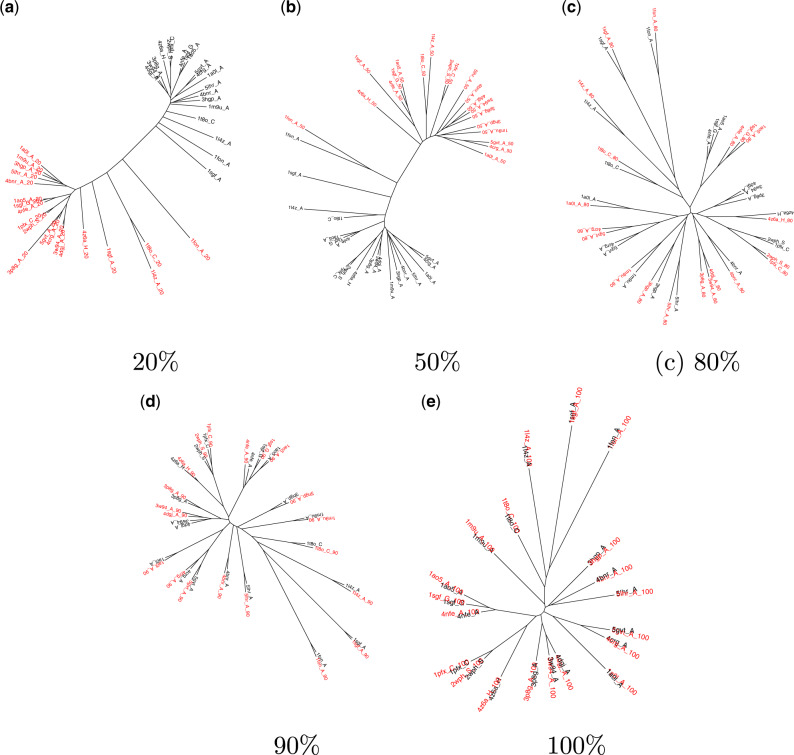
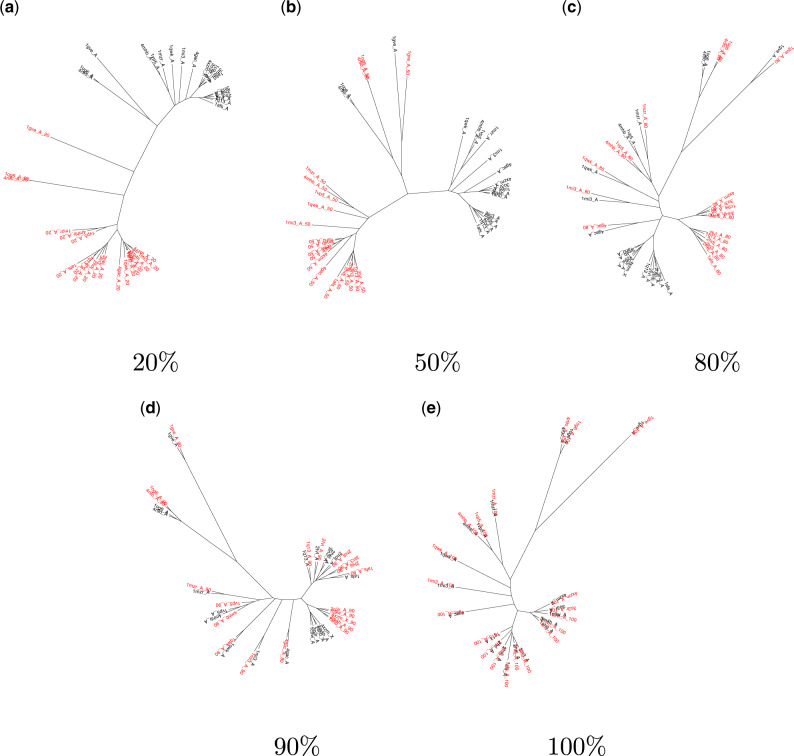
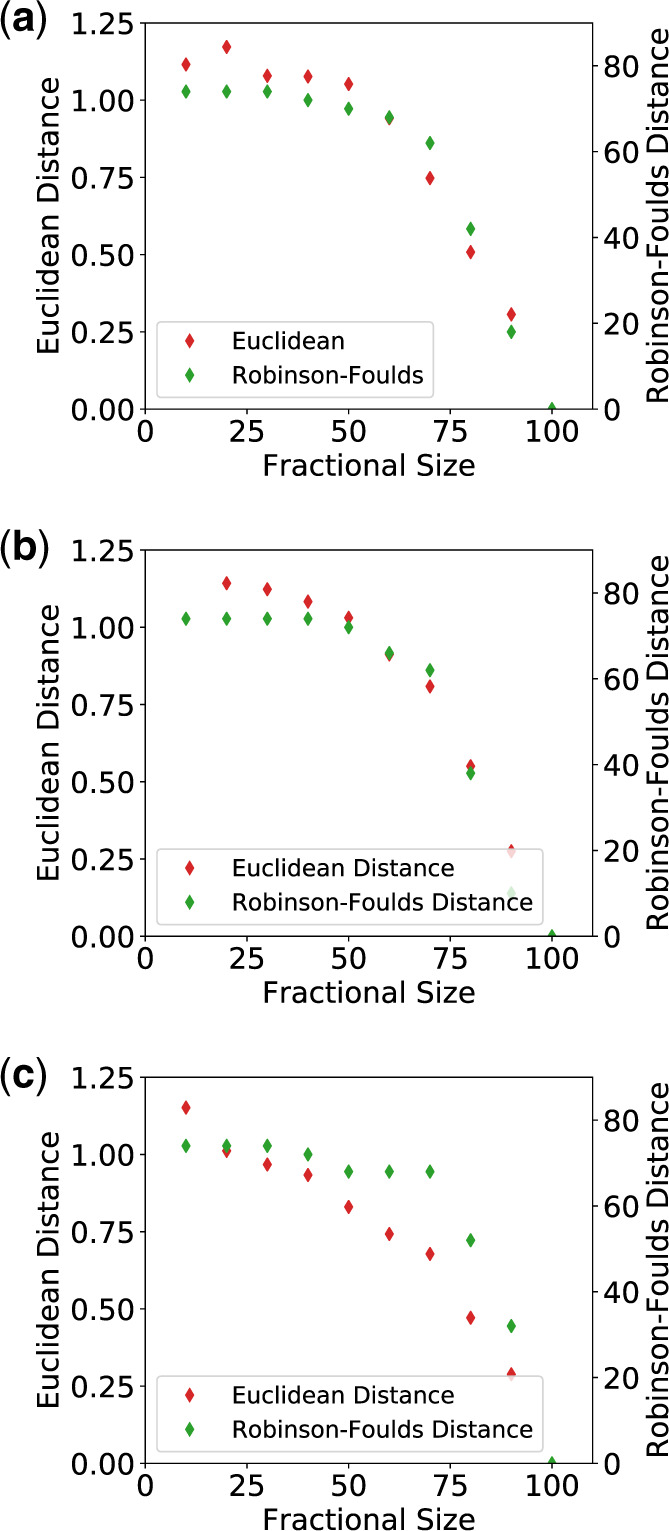
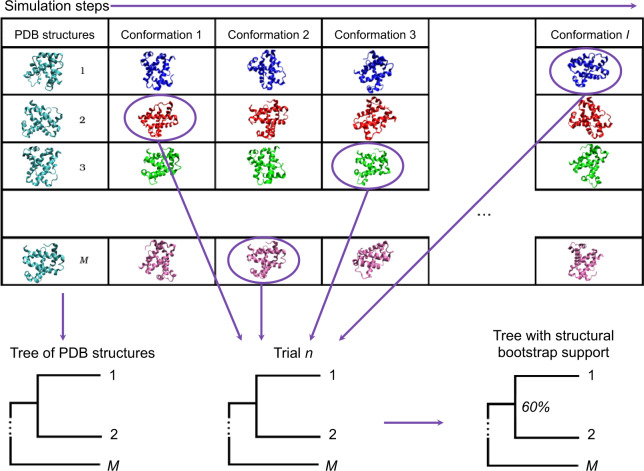
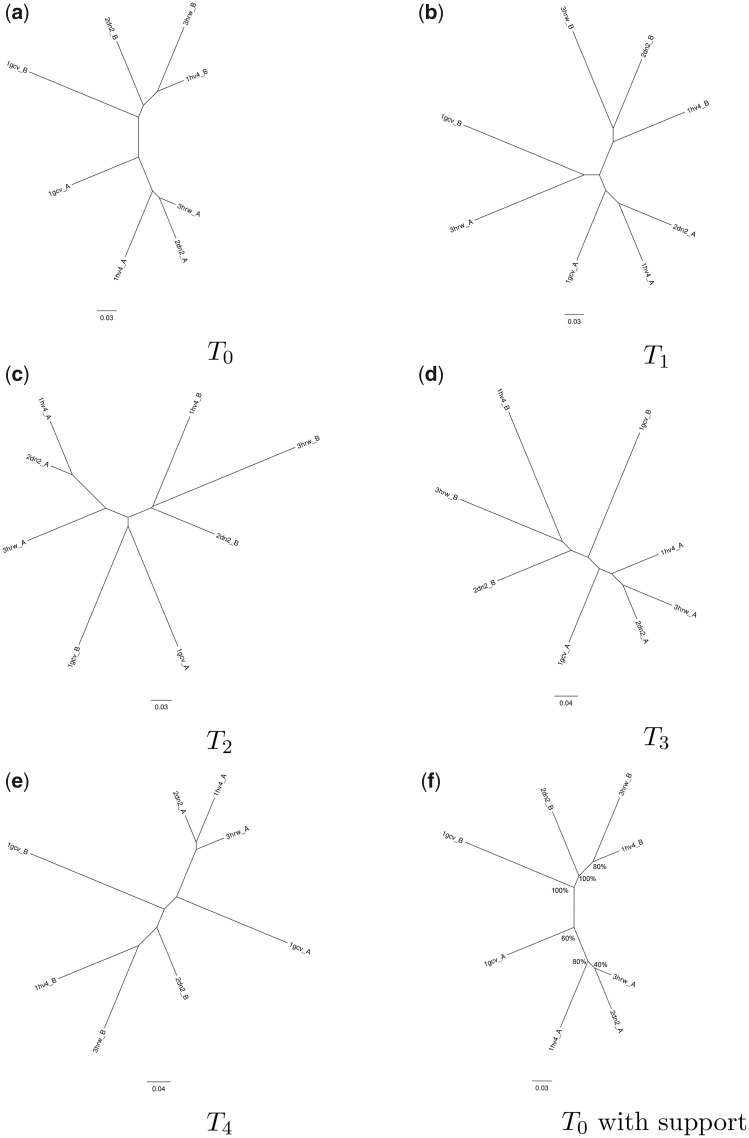
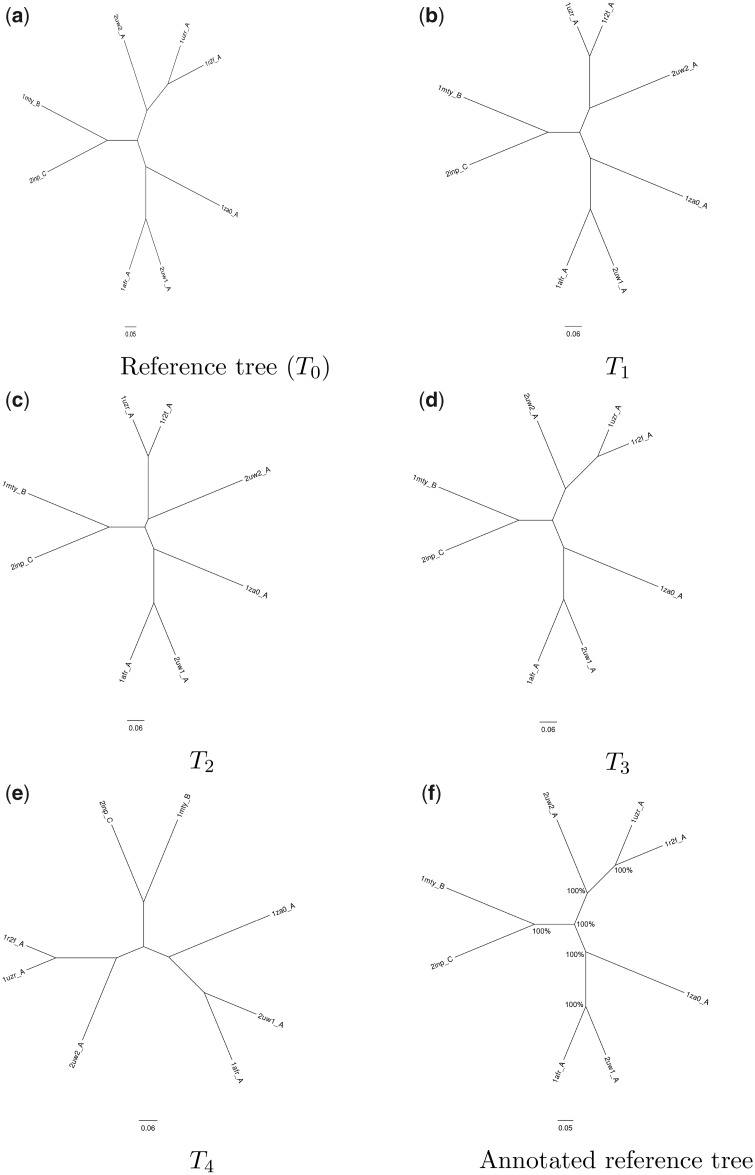
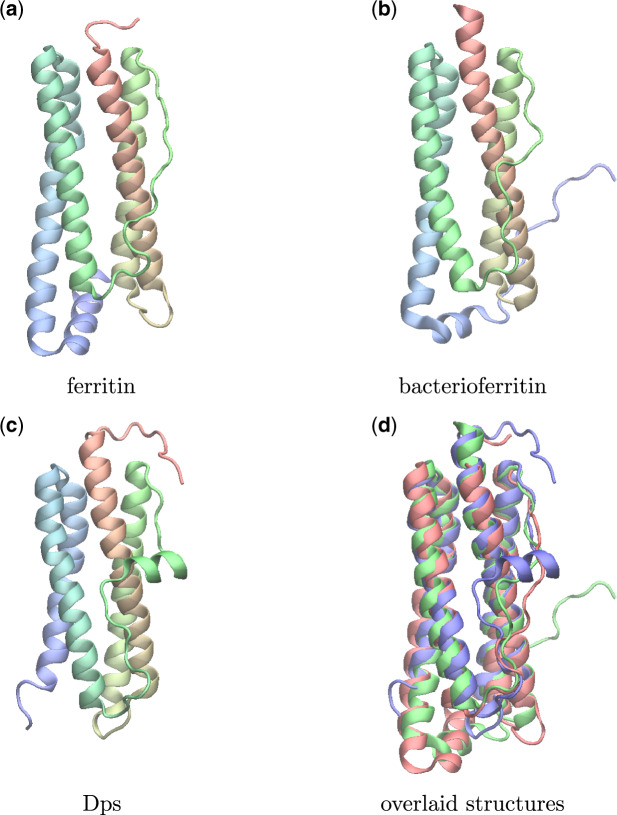
For evaluating the deepest evolutionary relationships among proteins, sequence similarity is too low for application of sequence-based homology search or phylogenetic methods. In such cases, comparison of protein structures, which are often better conserved than sequences, may provide an alternative means of uncovering deep evolutionary signal. Although major protein structure databases such as SCOP and CATH hierarchically group protein structures, they do not describe the specific evolutionary relationships within a hierarchical level. Structural phylogenies have the potential to fill this gap. However, it is difficult to assess evolutionary relationships derived from structural phylogenies without some means of assessing confidence in such trees. We therefore address two shortcomings in the application of structural data to deep phylogeny. First, we examine whether phylogenies derived from pairwise structural comparisons are sensitive to differences in protein length and shape. We find that structural phylogenetics is best employed where structures have very similar lengths, and that shape fluctuations generated during molecular dynamics simulations impact pairwise comparisons, but not so drastically as to eliminate evolutionary signal. Second, we address the absence of statistical support for structural phylogeny. We present a method for assessing confidence in a structural phylogeny using shape fluctuations generated via molecular dynamics or Monte Carlo simulations of proteins. Our approach will aid the evolutionary reconstruction of relationships across structurally defined protein superfamilies. With the Protein Data Bank now containing in excess of 158,000 entries (December 2019), we predict that structural phylogenetics will become a useful tool for ordering the protein universe.
为了评估蛋白质之间最深远的进化关系,序列相似性太低,无法应用基于序列的同源搜索或系统发育方法。在这种情况下,比较蛋白质结构可能是一种替代方法,可以揭示深层次的进化信号。尽管 SCOP 和 CATH 等主要蛋白质结构数据库对蛋白质结构进行了层次分组,但它们并没有描述层次结构内的特定进化关系。结构系统发育学具有填补这一空白的潜力。然而,如果没有某种方法来评估这些树的置信度,就很难评估从结构系统发育学中得出的进化关系。因此,我们解决了将结构数据应用于深度系统发育学的两个缺点。首先,我们检查从两两结构比较中得出的系统发育是否对蛋白质长度和形状的差异敏感。我们发现,结构系统发生学在结构非常相似的情况下最适用,并且分子动力学模拟中产生的形状波动会影响两两比较,但不会严重到消除进化信号。其次,我们解决了结构系统发育缺乏统计支持的问题。我们提出了一种使用通过分子动力学或蒙特卡罗模拟蛋白质生成的形状波动来评估结构系统发育置信度的方法。我们的方法将有助于在结构定义的蛋白质超家族中重建关系。随着蛋白质数据库现在包含超过 158,000 个条目(2019 年 12 月),我们预测结构系统发生学将成为一种有用的工具,用于对蛋白质宇宙进行排序。