Institute of Bioinformatics, University of Georgia, 120 Green Street, Athens, GA 30602, USA.
BMC Bioinformatics. 2012 Aug 21;13:209. doi: 10.1186/1471-2105-13-209.
Ongoing innovation in phylogenetics and evolutionary biology has been accompanied by a proliferation of software tools, data formats, analytical techniques and web servers. This brings with it the challenge of integrating phylogenetic and other related biological data found in a wide variety of formats, and underlines the need for reusable software that can read, manipulate and transform this information into the various forms required to build computational pipelines.
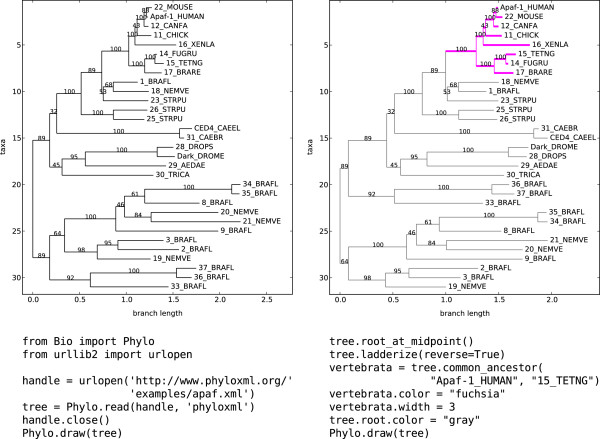
We built a Python software library for working with phylogenetic data that is tightly integrated with Biopython, a broad-ranging toolkit for computational biology. Our library, Bio.Phylo, is highly interoperable with existing libraries, tools and standards, and is capable of parsing common file formats for phylogenetic trees, performing basic transformations and manipulations, attaching rich annotations, and visualizing trees. We unified the modules for working with the standard file formats Newick, NEXUS and phyloXML behind a consistent and simple API, providing a common set of functionality independent of the data source.
Bio.Phylo meets a growing need in bioinformatics for working with heterogeneous types of phylogenetic data. By supporting interoperability with multiple file formats and leveraging existing Biopython features, this library simplifies the construction of phylogenetic workflows. We also provide examples of the benefits of building a community around a shared open-source project. Bio.Phylo is included with Biopython, available through the Biopython website, http://biopython.org.
系统发生学和进化生物学领域的持续创新,伴随着软件工具、数据格式、分析技术和网络服务器的大量涌现。这带来了整合以各种格式存储的系统发生学和其他相关生物学数据的挑战,也凸显了对可重复使用软件的需求,这种软件可以读取、操作和转换这些信息,以构建计算流程所需的各种形式。
我们构建了一个用于处理系统发生学数据的 Python 软件库,该库与计算生物学的广泛工具包 Biopython 紧密集成。我们的 Bio.Phylo 库与现有库、工具和标准高度互操作,能够解析常见的系统发生树文件格式,执行基本的转换和操作,附加丰富的注释,并可视化树。我们在一致且简单的 API 背后统一了用于处理 Newick、NEXUS 和 phyloXML 标准文件格式的模块,提供了一组独立于数据源的通用功能。
Bio.Phylo 满足了生物信息学领域对处理异构类型系统发生学数据的需求。通过支持与多种文件格式的互操作性并利用现有的 Biopython 功能,该库简化了系统发生工作流程的构建。我们还提供了围绕共享开源项目构建社区的好处的示例。Bio.Phylo 随 Biopython 一起提供,可通过 Biopython 网站获取,网址为 http://biopython.org。