Maharaj Avinaash, Theodorou Demetria, Banerjee Indraneel Indi, Metherell Louise A, Prasad Rathi, Wallace Dean
Centre for Endocrinology, John Vane Science Centre, William Harvey Research Institute, Queen Mary University of London, London, United Kingdom.
Department of Paediatric Nephrology, Royal Manchester Children's Hospital, Manchester, United Kingdom.
Front Pediatr. 2020 Apr 8;8:151. doi: 10.3389/fped.2020.00151. eCollection 2020.
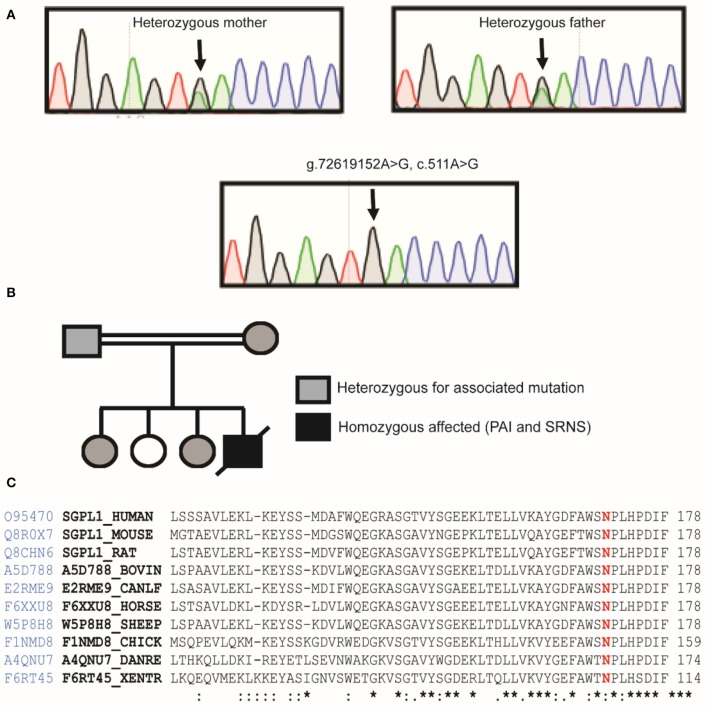
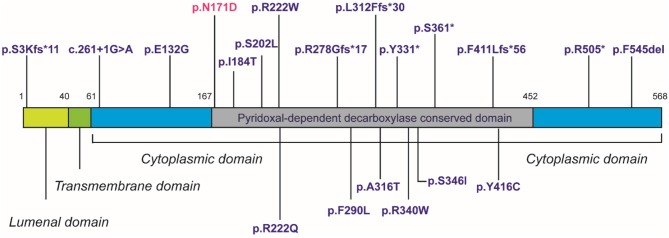
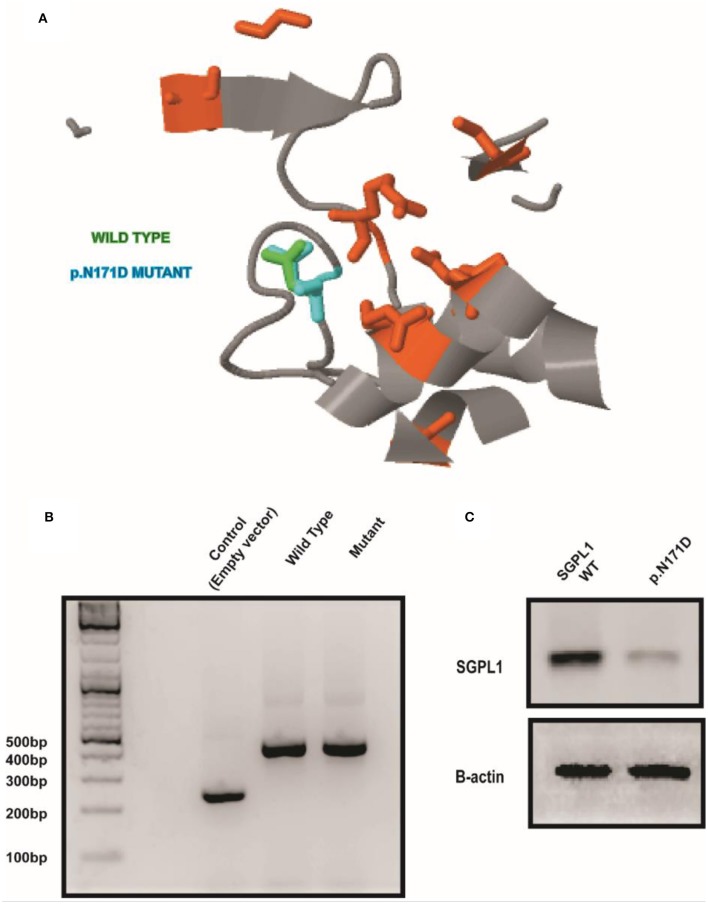
Loss of function mutations in are associated with Sphingosine-1-phosphate lyase insufficiency syndrome, comprising steroid resistant nephrotic syndrome, and primary adrenal insufficiency (PAI) in the majority of cases. encodes sphingosine-1-phosphate lyase (SGPL1) which is a major modulator of sphingolipid signaling. A Pakistani male infant presented at 5 months of age with failure to thrive, nephrotic syndrome, primary adrenal insufficiency, hypothyroidism, and hypogonadism. Other systemic manifestations included persistent lymphopenia, ichthyosis, and motor developmental delay. Aged 9 months, he progressed rapidly into end stage oligo-anuric renal failure and subsequently died. Sanger sequencing of the entire coding region of revealed the novel association of a rare homozygous mutation (chr10:72619152, c.511A>G, p.N171D; MAF-1.701e-05) with the condition. Protein expression of the p.N171D mutant was markedly reduced compared to wild type when overexpressed in an knockout cell line, and associated with a severe clinical phenotype. The case further highlights the emerging phenotype of patients with loss-of-function mutations. Whilst nephrotic syndrome is a recognized feature of other disorders of sphingolipid metabolism, sphingosine-1-phosphate lyase insufficiency syndrome is unique amongst the sphingolipidoses in presenting with multiple endocrinopathies. Given the multi-systemic and progressive nature of this form of PAI/ nephrotic syndrome, a genetic diagnosis is crucial for optimal management and appropriate screening for comorbidities in these patients.
[基因名称]功能丧失性突变与鞘氨醇-1-磷酸裂解酶不足综合征相关,在大多数情况下包括类固醇抵抗性肾病综合征和原发性肾上腺功能不全(PAI)。[基因名称]编码鞘氨醇-1-磷酸裂解酶(SGPL1),它是鞘脂信号传导的主要调节因子。一名巴基斯坦男婴在5个月大时出现发育不良、肾病综合征、原发性肾上腺功能不全、甲状腺功能减退和性腺功能减退。其他全身表现包括持续性淋巴细胞减少、鱼鳞病和运动发育迟缓。9个月大时,他迅速进展为终末期少尿性肾衰竭,随后死亡。对[基因名称]整个编码区进行桑格测序,发现一种罕见的纯合突变(chr10:72619152,c.511A>G,p.N171D;MAF-1.701e-05)与该病症存在新的关联。当在[基因名称]敲除细胞系中过表达时,p.N171D突变体的蛋白质表达与野生型相比明显降低,并与严重的临床表型相关。该病例进一步突出了功能丧失性[基因名称]突变患者的新出现的表型。虽然肾病综合征是其他鞘脂代谢紊乱的公认特征,但鞘氨醇-1-磷酸裂解酶不足综合征在鞘脂贮积病中独特之处在于伴有多种内分泌病。鉴于这种形式的PAI/肾病综合征具有多系统和进行性的性质,基因诊断对于这些患者的最佳管理和合并症的适当筛查至关重要。