Bioinformatics Research Center, North Carolina State University, Raleigh, North Carolina 27695
Department of Horticultural Science, North Carolina State University, Raleigh, North Carolina 27695.
Genetics. 2020 Jul;215(3):579-595. doi: 10.1534/genetics.120.303080. Epub 2020 May 5.
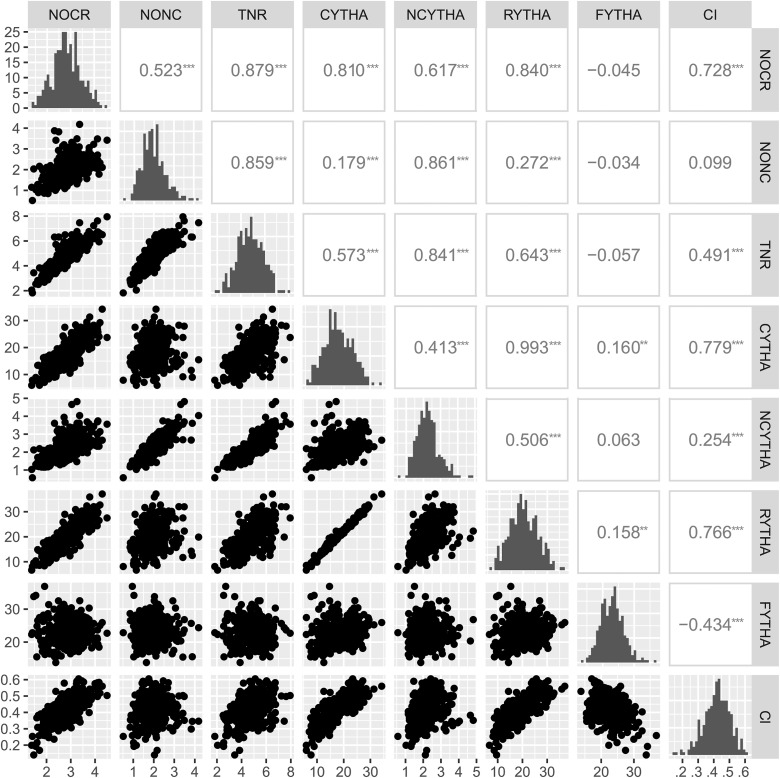
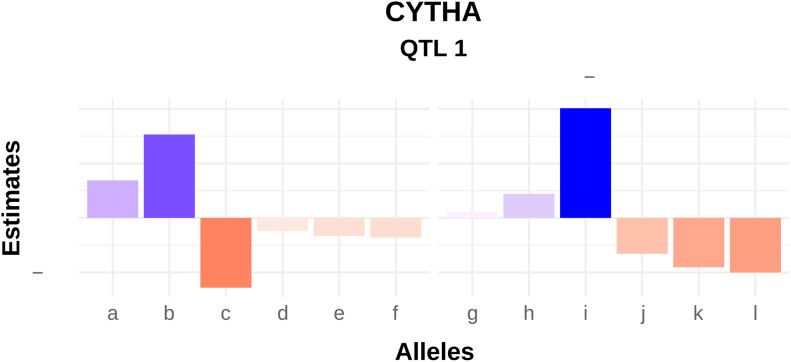
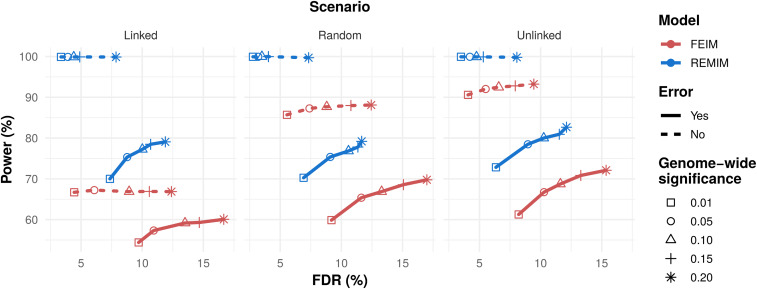
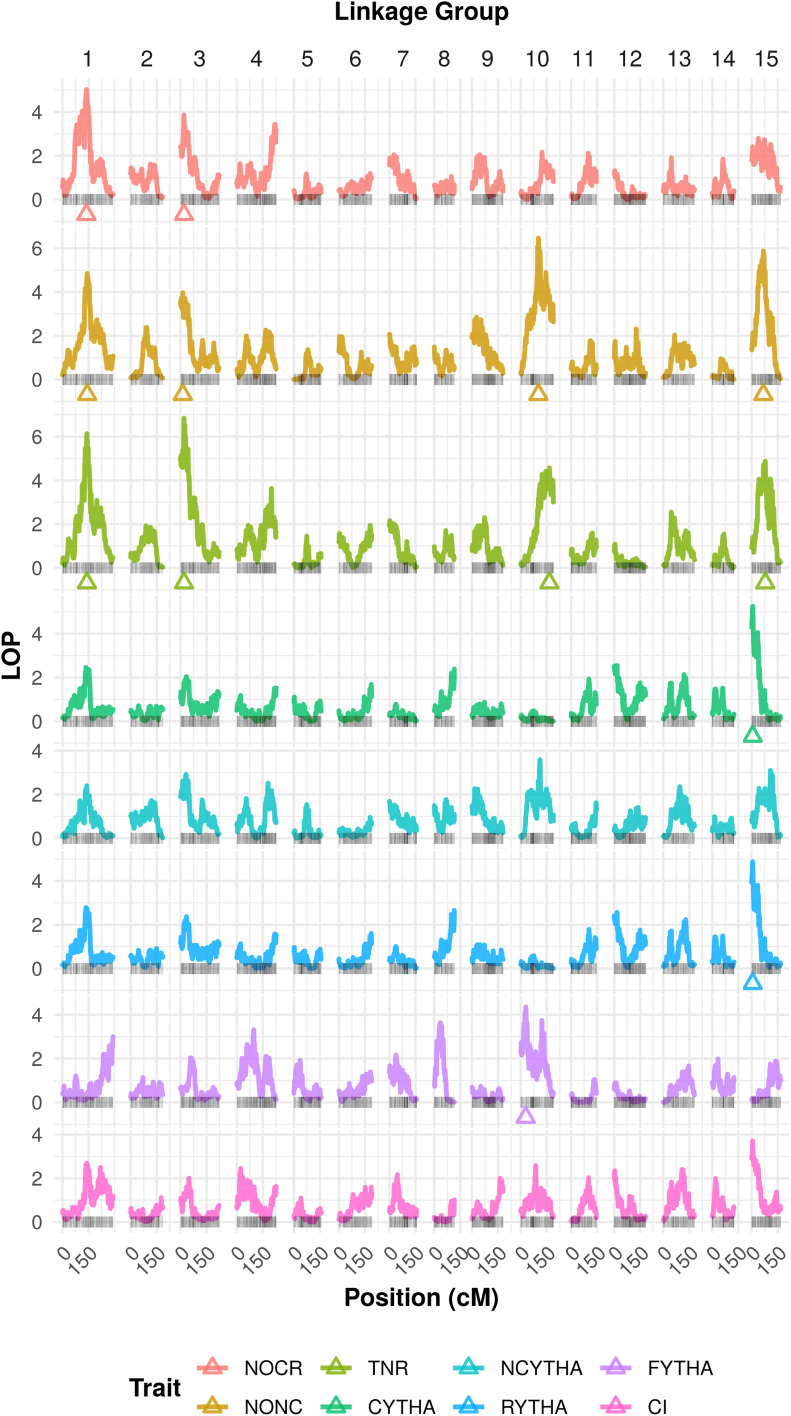
In developing countries, the sweetpotato, (L.) Lam. [Formula: see text], is an important autopolyploid species, both socially and economically. However, quantitative trait loci (QTL) mapping has remained limited due to its genetic complexity. Current fixed-effect models can fit only a single QTL and are generally hard to interpret. Here, we report the use of a random-effect model approach to map multiple QTL based on score statistics in a sweetpotato biparental population ('Beauregard' × 'Tanzania') with 315 full-sibs. Phenotypic data were collected for eight yield component traits in six environments in Peru, and jointly adjusted means were obtained using mixed-effect models. An integrated linkage map consisting of 30,684 markers distributed along 15 linkage groups (LGs) was used to obtain the genotype conditional probabilities of putative QTL at every centiMorgan position. Multiple interval mapping was performed using our R package QTLpoly and detected a total of 13 QTL, ranging from none to four QTL per trait, which explained up to 55% of the total variance. Some regions, such as those on LGs 3 and 15, were consistently detected among root number and yield traits, and provided a basis for candidate gene search. In addition, some QTL were found to affect commercial and noncommercial root traits distinctly. Further best linear unbiased predictions were decomposed into additive allele effects and were used to compute multiple QTL-based breeding values for selection. Together with quantitative genotyping and its appropriate usage in linkage analyses, this QTL mapping methodology will facilitate the use of genomic tools in sweetpotato breeding as well as in other autopolyploids.
在发展中国家,番薯(L.)Lam. [Formula: see text] 是一种重要的同源多倍体物种,在社会和经济方面都具有重要意义。然而,由于其遗传复杂性,数量性状位点(QTL)的作图一直受到限制。目前的固定效应模型只能拟合单个 QTL,并且通常难以解释。在这里,我们报告了使用基于得分统计的随机效应模型方法来映射基于‘Beauregard’בTanzania’双亲群体的多个 QTL,该群体共有 315 个全同胞。在秘鲁的六个环境中收集了八个产量构成性状的表型数据,并使用混合效应模型获得了联合调整均值。一个由 30684 个标记组成的整合连锁图谱,分布在 15 个连锁群(LG)上,用于在每个百分位位置获得假定 QTL 的基因型条件概率。使用我们的 R 包 QTLpoly 进行了多次区间作图,共检测到 13 个 QTL,每个性状从无到 4 个 QTL,解释了高达 55%的总方差。一些区域,如 LG3 和 LG15 上的区域,在根数和产量性状中始终被检测到,为候选基因搜索提供了基础。此外,一些 QTL 被发现对商业和非商业根性状有明显的影响。进一步的最佳线性无偏预测被分解为加性等位基因效应,并用于计算多个基于 QTL 的选择育种值。结合定量基因分型及其在连锁分析中的适当使用,这种 QTL 作图方法将促进基因组工具在番薯育种以及其他同源多倍体中的应用。