Medical Department, Genomi-k S.A.P.I. de C.V., Monterrey, Mexico.
Department of Medical Research, Cienciamed, Monterrey, Mexico.
Am J Case Rep. 2020 May 11;21:e919463. doi: 10.12659/AJCR.919463.
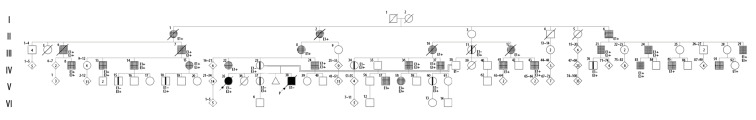
BACKGROUND Hereditary spastic paraplegia (HSP or SPG) consists of a heterogeneous group of disorders, clinically divided into pure and complex forms. The former is characterized by neurological impairment limited to lower-extremity spasticity. The latter presents additional symptoms such as seizures, psychomotor impairment, cataract, deafness, and peripheral neuropathy. The genetic structure of HSP is diverse, with more than 72 loci and 55 genes identified so far. The most common type is SPG4, accounting for 40% of cases. This case report describes 2 siblings presenting SPG4, one presumptive and one confirmed with a homozygous SPAST variant. CASE REPORT Two siblings born to third-degree consanguineous and healthy parents presented a SPG4 complex phenotype characterized by progressive psychomotor deterioration, mixed seizure patterns, corneal opacity, dysostotic bones, limb spasticity with extensor plantar responses, and axial hypotonia. After ruling out most inborn errors of metabolism in one of the patients, the complexity of the case derived from exome sequencing. The identification of a homozygous variant in the SPAST gene established a diagnosis for SPG4. The phenotype-genotype did not correlate to classical manifestations, most likely due to the variant's zygosity. Moreover, 34 patient's relatives were identified with SPG4 clinical manifestations or asymptomatic with the same genetic variant in heterozygous state. CONCLUSIONS We described visual loss and seizures for SPG4 complex phenotype associated with a homozygous variant in the SPAST gene. This diagnosis will lead clinicians to consider it as a differential diagnosis providing adequate genetic counseling.
背景 遗传性痉挛性截瘫(HSP 或 SPG)由一组异质性疾病组成,临床上分为单纯型和复杂型。前者的特点是神经系统损害仅限于下肢痉挛。后者表现出其他症状,如癫痫、精神运动障碍、白内障、耳聋和周围神经病。HSP 的遗传结构多种多样,迄今已确定超过 72 个位点和 55 个基因。最常见的类型是 SPG4,占病例的 40%。本病例报告描述了 2 名存在 SPG4 的兄弟姐妹,其中 1 名疑似,1 名经纯合 SPAST 变异确认。
病例报告 两名出生于三代近亲且健康的父母的兄弟姐妹表现出 SPG4 复杂表型,特征为进行性精神运动恶化、混合性癫痫发作模式、角膜混浊、骨发育不良、四肢痉挛伴伸性跖反射和轴性低张力。在排除了其中 1 名患者的大多数先天性代谢错误后,该病例的复杂性源自外显子组测序。在 SPAST 基因中发现一个纯合变异,从而确立了 SPG4 的诊断。表型-基因型与经典表现不相关,最有可能是由于变异的纯合性。此外,还鉴定了 34 名患者的亲属存在 SPG4 临床表现或无症状,其携带相同的杂合遗传变异。
结论 我们描述了与 SPAST 基因纯合变异相关的 SPG4 复杂表型的视力丧失和癫痫发作。这种诊断将促使临床医生将其作为一种鉴别诊断,提供充分的遗传咨询。