School of Life Sciences, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015, Lausanne, Switzerland.
Swiss Institute of Bioinformatics (SIB), CH-1015, Lausanne, Switzerland.
Genome Biol. 2020 May 11;21(1):114. doi: 10.1186/s13059-020-01996-3.
Positional weight matrix (PWM) is a de facto standard model to describe transcription factor (TF) DNA binding specificities. PWMs inferred from in vivo or in vitro data are stored in many databases and used in a plethora of biological applications. This calls for comprehensive benchmarking of public PWM models with large experimental reference sets.
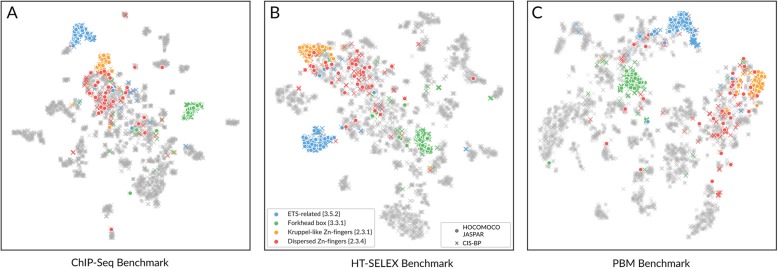
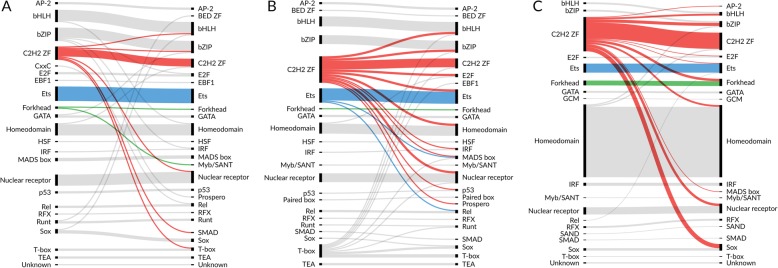
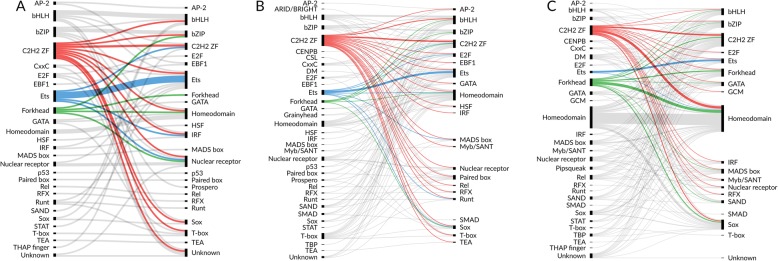
Here we report results from all-against-all benchmarking of PWM models for DNA binding sites of human TFs on a large compilation of in vitro (HT-SELEX, PBM) and in vivo (ChIP-seq) binding data. We observe that the best performing PWM for a given TF often belongs to another TF, usually from the same family. Occasionally, binding specificity is correlated with the structural class of the DNA binding domain, indicated by good cross-family performance measures. Benchmarking-based selection of family-representative motifs is more effective than motif clustering-based approaches. Overall, there is good agreement between in vitro and in vivo performance measures. However, for some in vivo experiments, the best performing PWM is assigned to an unrelated TF, indicating a binding mode involving protein-protein cooperativity.
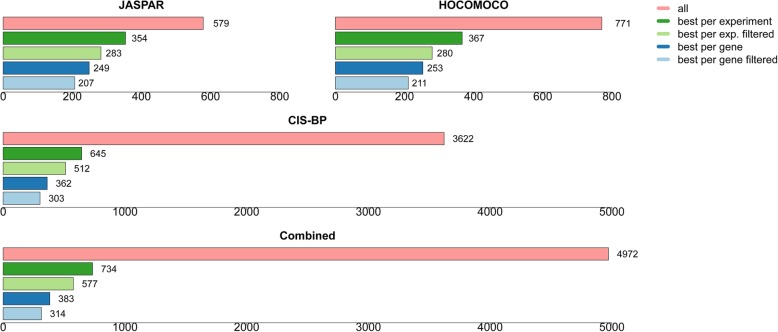
In an all-against-all setting, we compute more than 18 million performance measure values for different PWM-experiment combinations and offer these results as a public resource to the research community. The benchmarking protocols are provided via a web interface and as docker images. The methods and results from this study may help others make better use of public TF specificity models, as well as public TF binding data sets.
位置权重矩阵(PWM)是描述转录因子(TF)DNA 结合特异性的事实上的标准模型。从体内或体外数据推断出的 PWMs 存储在许多数据库中,并在众多生物学应用中使用。这就需要用大型实验参考集对公共 PWM 模型进行全面基准测试。
我们在此报告了对人类 TF 的 DNA 结合位点的 PWM 模型进行全面比较的结果,这些模型是基于大量的体外(HT-SELEX、PBM)和体内(ChIP-seq)结合数据编译而成的。我们观察到,对于给定的 TF,表现最好的 PWM 通常属于另一个 TF,通常来自同一家族。偶尔,DNA 结合域的结构类别与结合特异性相关,这表明跨家族的性能度量值较好。基于基准测试的家族代表性基序选择比基于基序聚类的方法更有效。总体而言,体外和体内性能度量值之间有很好的一致性。然而,对于一些体内实验,表现最好的 PWM 被分配给一个不相关的 TF,这表明存在涉及蛋白质-蛋白质协同作用的结合模式。
在全面比较的情况下,我们计算了不同 PWM-实验组合的超过 1800 万个性能度量值,并将这些结果作为公共资源提供给研究社区。基准测试协议通过 Web 界面和 Docker 映像提供。本研究的方法和结果可以帮助其他人更好地利用公共 TF 特异性模型和公共 TF 结合数据集。