Bangayan Nathanael J, Shi Baochen, Trinh Jerry, Barnard Emma, Kasimatis Gabriela, Curd Emily, Li Huiying
Department of Molecular and Medical Pharmacology, Crump Institute for Molecular Imaging, University of California, Los Angeles, CA 90095, USA.
Department of Ecology and Evolutionary Biology, University of California, Los Angeles, CA 90095, USA.
Microorganisms. 2020 May 8;8(5):684. doi: 10.3390/microorganisms8050684.
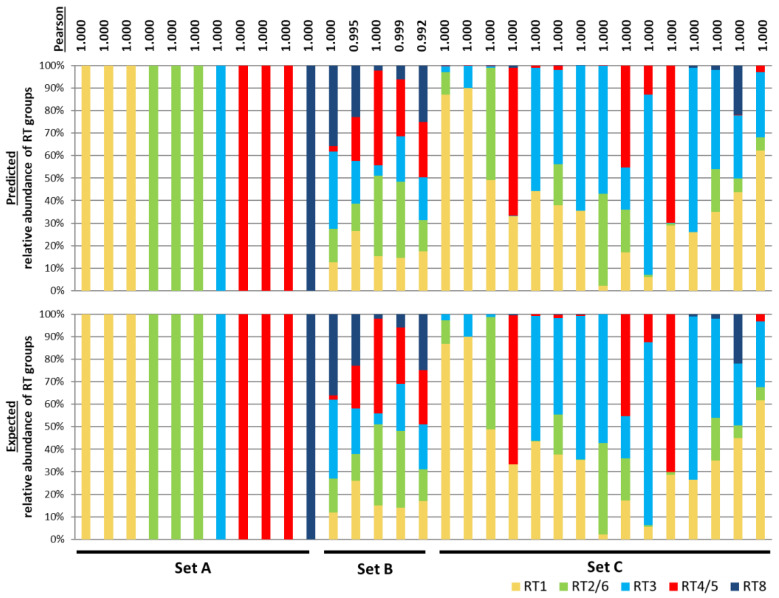
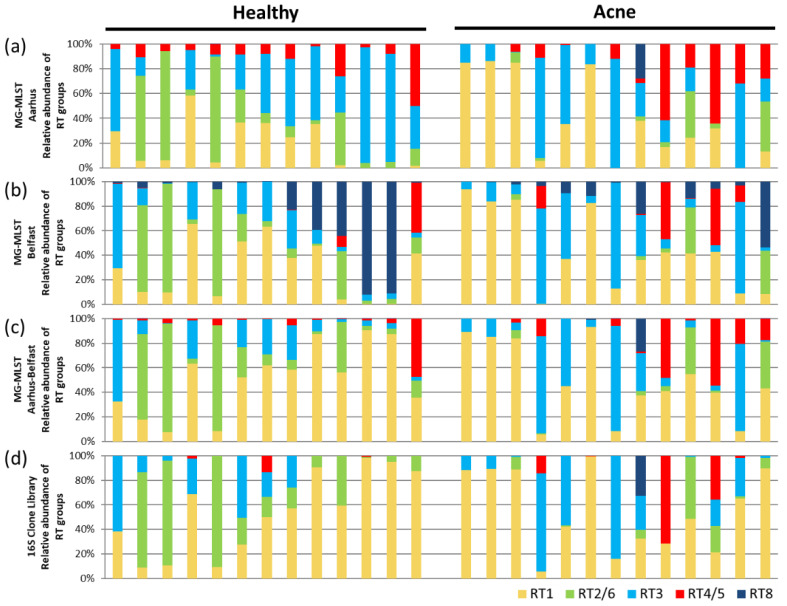
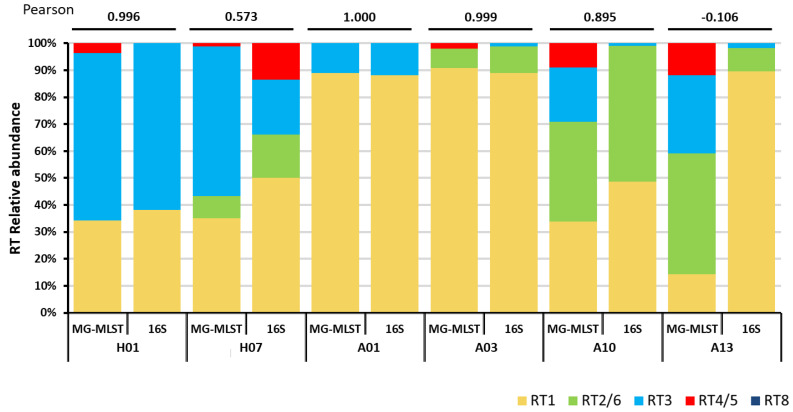
The microbiome plays an important role in human physiology. The composition of the human microbiome has been described at the phylum, class, genus, and species levels, however, it is largely unknown at the strain level. The importance of strain-level differences in microbial communities has been increasingly recognized in understanding disease associations. Current methods for identifying strain populations often require deep metagenomic sequencing and a comprehensive set of reference genomes. In this study, we developed a method, metagenomic multi-locus sequence typing (MG-MLST), to determine strain-level composition in a microbial community by combining high-throughput sequencing with multi-locus sequence typing (MLST). We used a commensal bacterium, , as an example to test the ability of MG-MLST in identifying the strain composition. Using simulated communities, MG-MLST accurately predicted the strain populations in all samples. We further validated the method using MLST gene amplicon libraries and metagenomic shotgun sequencing data of clinical skin samples. MG-MLST yielded consistent results of the strain composition to those obtained from nearly full-length 16S rRNA clone libraries and metagenomic shotgun sequencing analysis. When comparing strain-level differences between acne and healthy skin microbiomes, we demonstrated that strains of RT2/6 were highly associated with healthy skin, consistent with previous findings. In summary, MG-MLST provides a quantitative analysis of the strain populations in the microbiome with diversity and richness. It can be applied to microbiome studies to reveal strain-level differences between groups, which are critical in many microorganism-related diseases.
微生物群落在人体生理学中发挥着重要作用。人类微生物群落的组成已在门、纲、属和种水平上进行了描述,然而,在菌株水平上,其情况 largely unknown(此处原文有误,推测可能是“很大程度上未知”)。微生物群落中菌株水平差异的重要性在理解疾病关联方面已得到越来越多的认可。当前识别菌株群体的方法通常需要深度宏基因组测序和一套完整的参考基因组。在本研究中,我们开发了一种方法,即宏基因组多位点序列分型(MG-MLST),通过将高通量测序与多位点序列分型(MLST)相结合来确定微生物群落中的菌株水平组成。我们以一种共生细菌 为例来测试MG-MLST识别菌株组成的能力。使用模拟群落,MG-MLST准确预测了所有样本中的菌株群体。我们进一步使用MLST基因扩增子文库和临床皮肤样本的宏基因组鸟枪法测序数据对该方法进行了验证。MG-MLST得出的菌株组成结果与从近乎全长的16S rRNA克隆文库和宏基因组鸟枪法测序分析中获得的结果一致。在比较痤疮和健康皮肤微生物群之间的菌株水平差异时,我们证明RT2/6菌株与健康皮肤高度相关,这与先前的研究结果一致。总之,MG-MLST提供了对微生物群落中菌株群体的多样性和丰富度的定量分析。它可应用于微生物群落研究,以揭示不同群体之间的菌株水平差异,这在许多与微生物相关的疾病中至关重要。