Oklahoma Center for Neuroscience, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma.
Department of Basic Science and Craniofacial Biology, New York University, New York City, New York.
Cell Mol Gastroenterol Hepatol. 2020;10(3):527-543. doi: 10.1016/j.jcmgh.2020.04.020. Epub 2020 May 8.
BACKGROUND & AIMS: Psychological stress is a trigger for the development of irritable bowel syndrome and associated symptoms including abdominal pain. Although irritable bowel syndrome patients show increased activation in the limbic brain, including the amygdala, the underlying molecular and cellular mechanisms regulating visceral nociception in the central nervous system are incompletely understood. In a rodent model of chronic stress, we explored the role of microglia in the central nucleus of the amygdala (CeA) in controlling visceral sensitivity. Microglia are activated by environmental challenges such as stress, and are able to modify neuronal activity via synaptic remodeling and inflammatory cytokine release. Inflammatory gene expression and microglial activity are regulated negatively by nuclear glucocorticoid receptors (GR), which are suppressed by the stress-activated pain mediator p38 mitogen-activated protein kinases (MAPK).
Fisher-344 male rats were exposed to water avoidance stress (WAS) for 1 hour per day for 7 days. Microglia morphology and the expression of phospho-p38 MAPK and GR were analyzed via immunofluorescence. Microglia-mediated synaptic remodeling was investigated by quantifying the number of postsynaptic density protein 95-positive puncta. Cytokine expression levels in the CeA were assessed via quantitative polymerase chain reaction and a Luminex assay (Bio-Rad, Hercules, CA). Stereotaxic infusion into the CeA of minocycline to inhibit, or fractalkine to activate, microglia was followed by colonic sensitivity measurement via a visceromotor behavioral response to isobaric graded pressures of tonic colorectal distension.
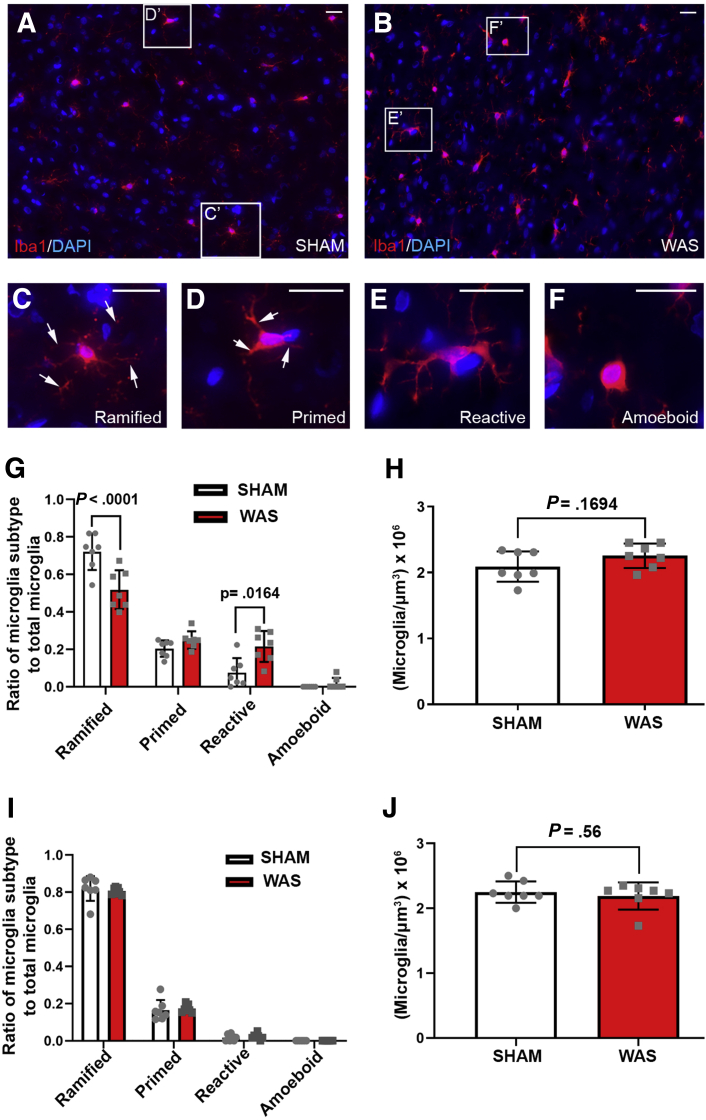
WAS induced microglial deramification in the CeA. Moreover, WAS induced a 3-fold increase in the expression of phospho-p38 and decreased the ratio of nuclear GR in the microglia. The number of microglia-engulfed postsynaptic density protein 95-positive puncta in the CeA was increased 3-fold by WAS, while cytokine levels were unchanged. WAS-induced changes in microglial morphology, microglia-mediated synaptic engulfment in the CeA, and visceral hypersensitivity were reversed by minocycline whereas in stress-naïve rats, fractalkine induced microglial deramification and visceral hypersensitivity.
Our data show that chronic stress induces visceral hypersensitivity in male rats and is associated with microglial p38 MAPK activation, GR dysfunction, and neuronal remodeling in the CeA.
心理压力是肠易激综合征和相关症状(包括腹痛)发展的一个诱因。虽然肠易激综合征患者的边缘大脑(包括杏仁核)表现出更高的激活,但调节中枢神经系统内脏痛觉的潜在分子和细胞机制尚不完全清楚。在慢性应激的啮齿动物模型中,我们探索了中央杏仁核(CeA)中的小胶质细胞在控制内脏敏感性中的作用。小胶质细胞受到环境挑战(如压力)的激活,能够通过突触重塑和炎性细胞因子释放来改变神经元活性。核糖皮质激素受体(GR)负调节炎性基因表达和小胶质细胞活性,而应激激活的疼痛介体 p38 丝裂原活化蛋白激酶(MAPK)抑制 GR。
将 Fisher-344 雄性大鼠暴露于每天 1 小时的水回避应激(WAS)中,持续 7 天。通过免疫荧光分析小胶质细胞形态以及磷酸化 p38 MAPK 和 GR 的表达。通过量化突触后密度蛋白 95 阳性斑点的数量来研究小胶质细胞介导的突触重塑。通过定量聚合酶链反应和 Luminex 测定(Bio-Rad,加利福尼亚州赫拉克勒斯)评估 CeA 中的细胞因子表达水平。立体定向 CeA 内注射米诺环素抑制小胶质细胞或 fractalkine 激活小胶质细胞,然后通过等渗分级压力下的肠道运动行为反应测量结肠敏感性。
WAS 诱导 CeA 中小胶质细胞的脱分支。此外,WAS 诱导磷酸化 p38 的表达增加 3 倍,并降低 CeA 中小胶质细胞中核 GR 的比例。WAS 使 CeA 中被小胶质细胞吞噬的突触后密度蛋白 95 阳性斑点的数量增加了 3 倍,而细胞因子水平不变。米诺环素逆转了 WAS 诱导的小胶质细胞形态、CeA 中小胶质细胞介导的突触吞噬作用和内脏高敏性的改变,而在应激-naive 大鼠中, fractalkine 诱导小胶质细胞脱分支和内脏高敏性。
我们的数据表明,慢性应激诱导雄性大鼠内脏高敏,并与 CeA 中小胶质细胞 p38 MAPK 激活、GR 功能障碍和神经元重塑有关。