Maranhão Andréa Queiroz, Silva Heidi Muniz, da Silva Waldeyr Mendes Cordeiro, França Renato Kaylan Alves, De Leo Thais Canassa, Dias-Baruffi Marcelo, Burtet Rafael Trindade, Brigido Marcelo Macedo
Department of Cellular Biology, Institute of Biological Science, University of Brasília, Brasília, Brazil.
Instituto de Investigação em Imunologia, Instituto Nacional de Ciência e Tecnologia (iii-INCT), São Paulo, Brazil.
Bioinform Biol Insights. 2020 Apr 30;14:1177932220915240. doi: 10.1177/1177932220915240. eCollection 2020.
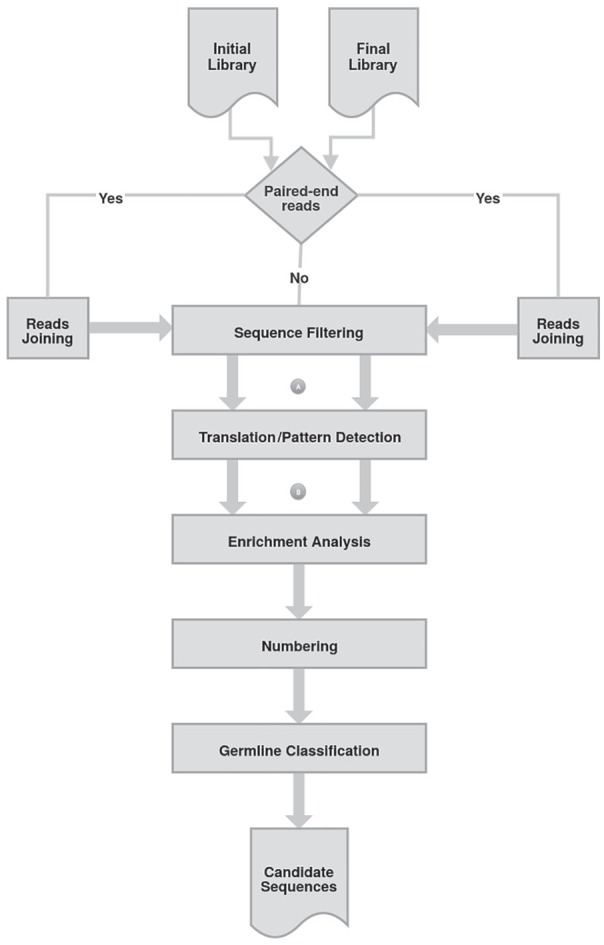
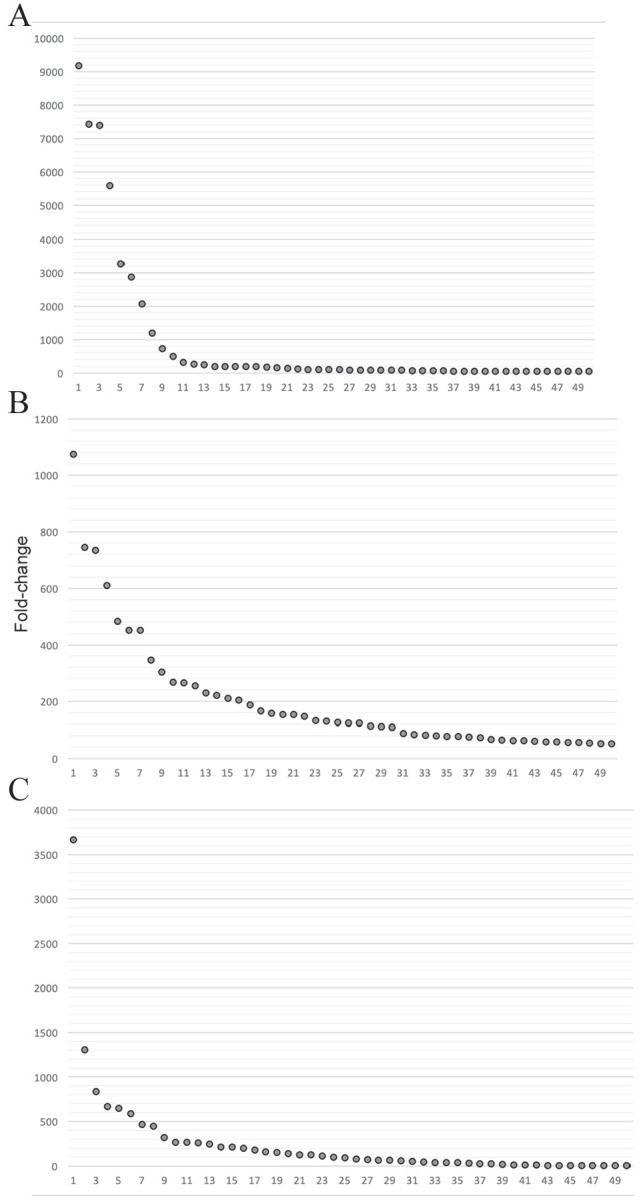
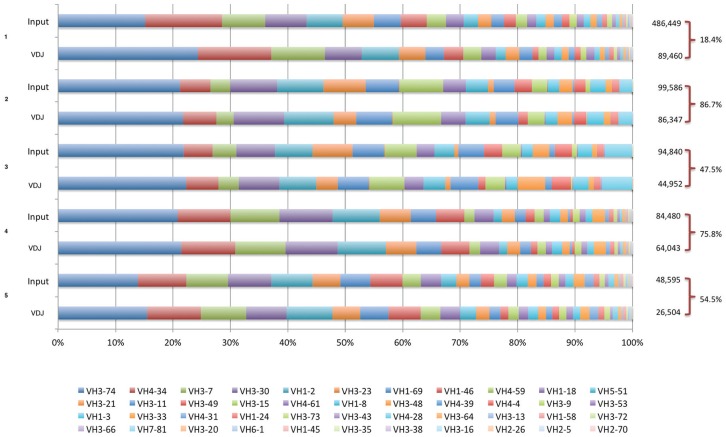
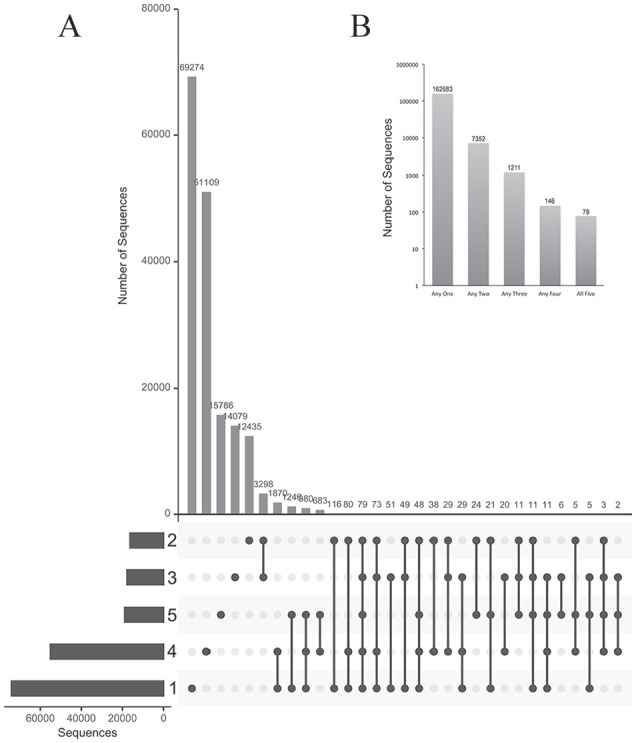
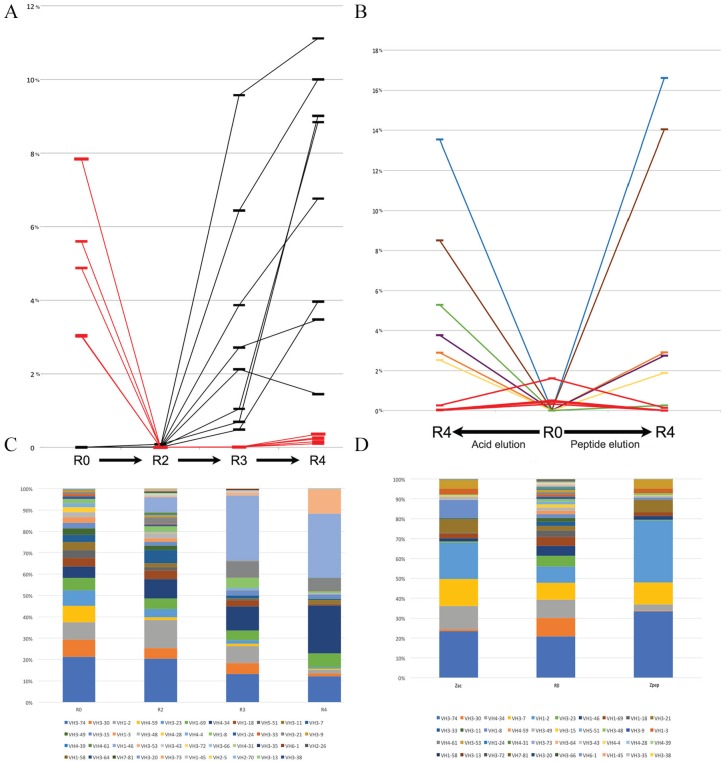
Phage display is a powerful technique to select high-affinity antibodies for different purposes, including biopharmaceuticals. Next-generation sequencing (NGS) presented itself as a robust solution, making it possible to assess billions of sequences of the variable domains from selected sublibraries. Handling this process, a central difficulty is to find the selected clones. Here, we present the AutomaTed Tool For Immunoglobulin Analysis (ATTILA), a new tool to analyze and find the enriched variable domains throughout a biopanning experiment. The ATTILA is a workflow that combines publicly available tools and in-house programs and scripts to find the fold-change frequency of deeply sequenced amplicons generated from selected VH and VL domains. We analyzed the same human Fab library NGS data using ATTILA in 5 different experiments, as well as on 2 biopanning experiments regarding performance, accuracy, and output. These analyses proved to be suitable to assess library variability and to list the more enriched variable domains, as ATTILA provides a report with the amino acid sequence of each identified domain, along with its complementarity-determining regions (CDRs), germline classification, and fold change. Finally, the methods employed here demonstrated a suitable manner to combine amplicon generation and NGS data analysis to discover new monoclonal antibodies (mAbs).
噬菌体展示是一种强大的技术,可用于为包括生物制药在内的不同目的筛选高亲和力抗体。新一代测序(NGS)是一种强大的解决方案,它使评估来自选定亚文库的数十亿可变区序列成为可能。在处理这个过程时,一个核心难题是找到选定的克隆。在此,我们介绍了免疫球蛋白分析自动化工具(ATTILA),这是一种用于在整个生物淘选实验中分析和找到富集可变区的新工具。ATTILA是一个工作流程,它结合了公开可用的工具以及内部程序和脚本,以找到从选定的重链可变区(VH)和轻链可变区(VL)生成的深度测序扩增子的倍数变化频率。我们在5个不同的实验中使用ATTILA分析了相同的人源Fab文库NGS数据,以及在2个关于性能、准确性和输出的生物淘选实验中进行了分析。这些分析被证明适用于评估文库变异性并列出更富集的可变区,因为ATTILA会提供一份报告,其中包含每个鉴定出的结构域的氨基酸序列,以及其互补决定区(CDR)、种系分类和倍数变化。最后,这里采用的方法展示了一种将扩增子生成与NGS数据分析相结合以发现新单克隆抗体(mAb)的合适方式。