Università degli Studi di Milano, AnacletoLab - Dipartimento di Informatica, via Giovanni Celoria 18, 20135 Milano, Italy.
Berlin Institute of Health (BIH), Anna-Louisa-Karsch-Straße 2, 10178 Berlin, Germany.
Gigascience. 2020 May 1;9(5). doi: 10.1093/gigascience/giaa052.
Several prediction problems in computational biology and genomic medicine are characterized by both big data as well as a high imbalance between examples to be learned, whereby positive examples can represent a tiny minority with respect to negative examples. For instance, deleterious or pathogenic variants are overwhelmed by the sea of neutral variants in the non-coding regions of the genome: thus, the prediction of deleterious variants is a challenging, highly imbalanced classification problem, and classical prediction tools fail to detect the rare pathogenic examples among the huge amount of neutral variants or undergo severe restrictions in managing big genomic data.
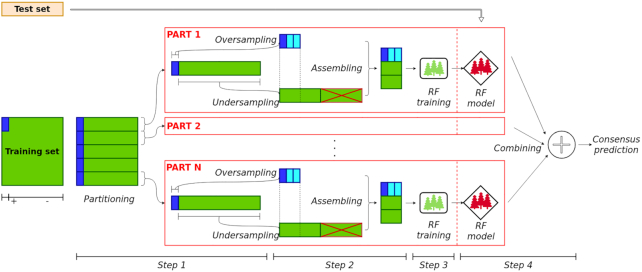
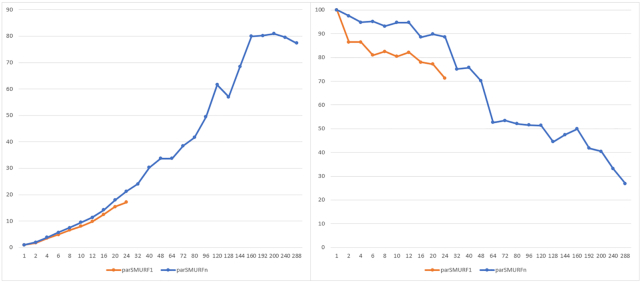
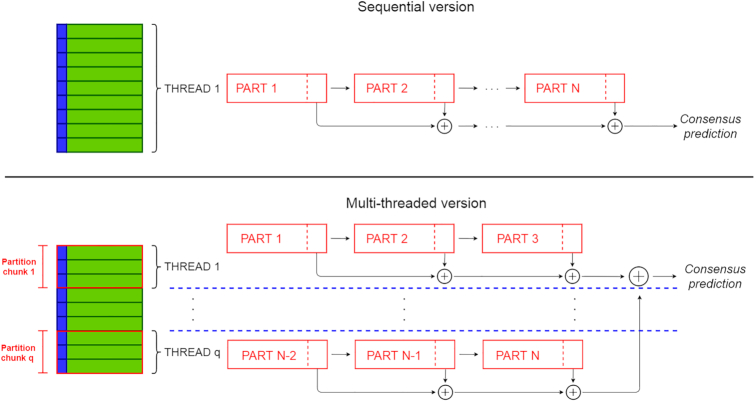
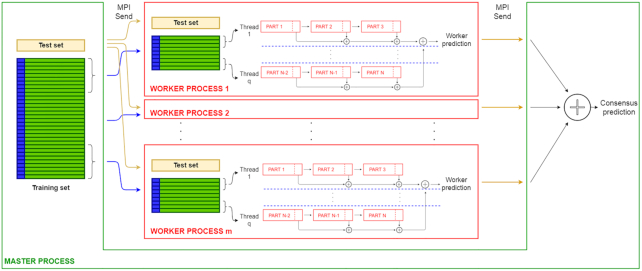
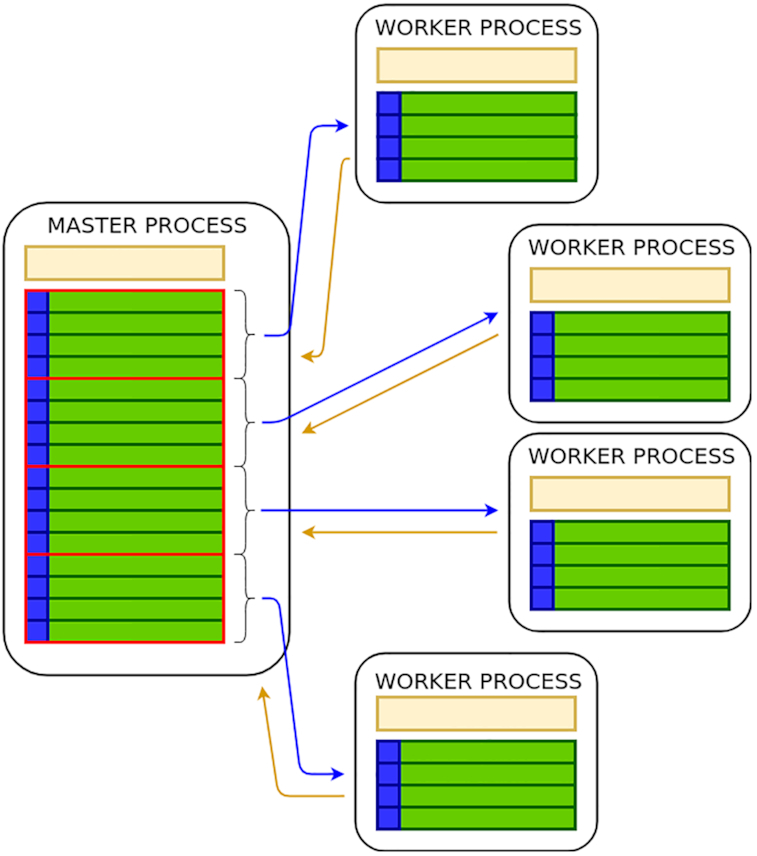
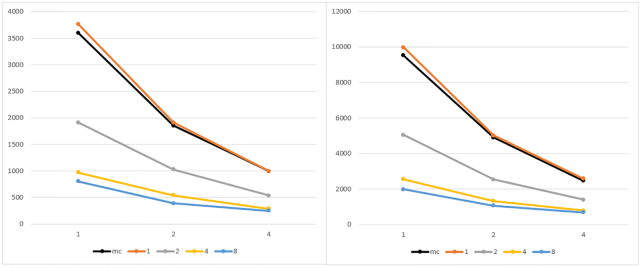
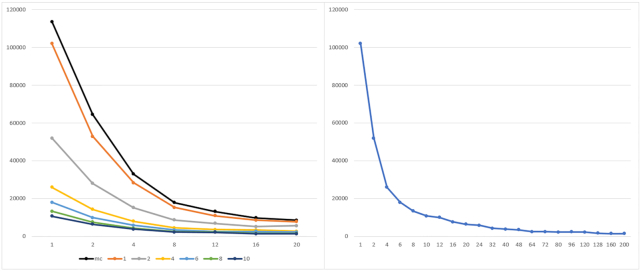
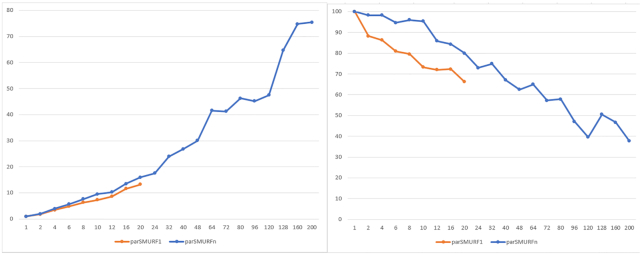
To overcome these limitations we propose parSMURF, a method that adopts a hyper-ensemble approach and oversampling and undersampling techniques to deal with imbalanced data, and parallel computational techniques to both manage big genomic data and substantially speed up the computation. The synergy between Bayesian optimization techniques and the parallel nature of parSMURF enables efficient and user-friendly automatic tuning of the hyper-parameters of the algorithm, and allows specific learning problems in genomic medicine to be easily fit. Moreover, by using MPI parallel and machine learning ensemble techniques, parSMURF can manage big data by partitioning them across the nodes of a high-performance computing cluster. Results with synthetic data and with single-nucleotide variants associated with Mendelian diseases and with genome-wide association study hits in the non-coding regions of the human genome, involhing millions of examples, show that parSMURF achieves state-of-the-art results and an 80-fold speed-up with respect to the sequential version.
parSMURF is a parallel machine learning tool that can be trained to learn different genomic problems, and its multiple levels of parallelization and high scalability allow us to efficiently fit problems characterized by big and imbalanced genomic data. The C++ OpenMP multi-core version tailored to a single workstation and the C++ MPI/OpenMP hybrid multi-core and multi-node parSMURF version tailored to a High Performance Computing cluster are both available at https://github.com/AnacletoLAB/parSMURF.
计算生物学和基因组医学中的几个预测问题都具有大数据和学习样本之间高度不平衡的特点,其中阳性样本相对于阴性样本可以代表很小的一部分。例如,在基因组的非编码区域中,有害或致病变体被大量中性变体所淹没:因此,有害变体的预测是一个具有挑战性的、高度不平衡的分类问题,传统的预测工具无法在大量中性变体中检测到罕见的致病实例,或者在处理大型基因组数据时受到严重限制。
为了克服这些限制,我们提出了 parSMURF 方法,该方法采用超集成方法和过采样和欠采样技术来处理不平衡数据,以及并行计算技术来管理大型基因组数据并大大加快计算速度。贝叶斯优化技术和 parSMURF 的并行性之间的协同作用使算法的超参数的高效和用户友好的自动调整成为可能,并允许对基因组医学中的特定学习问题进行轻松拟合。此外,通过使用 MPI 并行和机器学习集成技术,parSMURF 可以通过将大数据分割到高性能计算集群的节点上来管理大数据。使用合成数据以及与单核苷酸变体相关的孟德尔疾病和人类基因组非编码区域中的全基因组关联研究命中的结果,涉及数百万个实例,表明 parSMURF 达到了最先进的结果,并与顺序版本相比实现了 80 倍的加速。
parSMURF 是一种并行机器学习工具,可以针对不同的基因组问题进行训练,其多层次的并行化和高度可扩展性允许我们有效地拟合具有大数据和不平衡基因组数据的问题。针对单个工作站的 C++ OpenMP 多核版本和针对高性能计算集群的 C++ MPI/OpenMP 混合多核和多节点 parSMURF 版本都可以在 https://github.com/AnacletoLAB/parSMURF 上获得。