Nakagawa Hidewaki, Fujita Masashi
Laboratory for Genome Sequencing Analysis, RIKEN Center for Integrative Medical Sciences, Tokyo, Japan.
Cancer Sci. 2018 Mar;109(3):513-522. doi: 10.1111/cas.13505. Epub 2018 Feb 26.
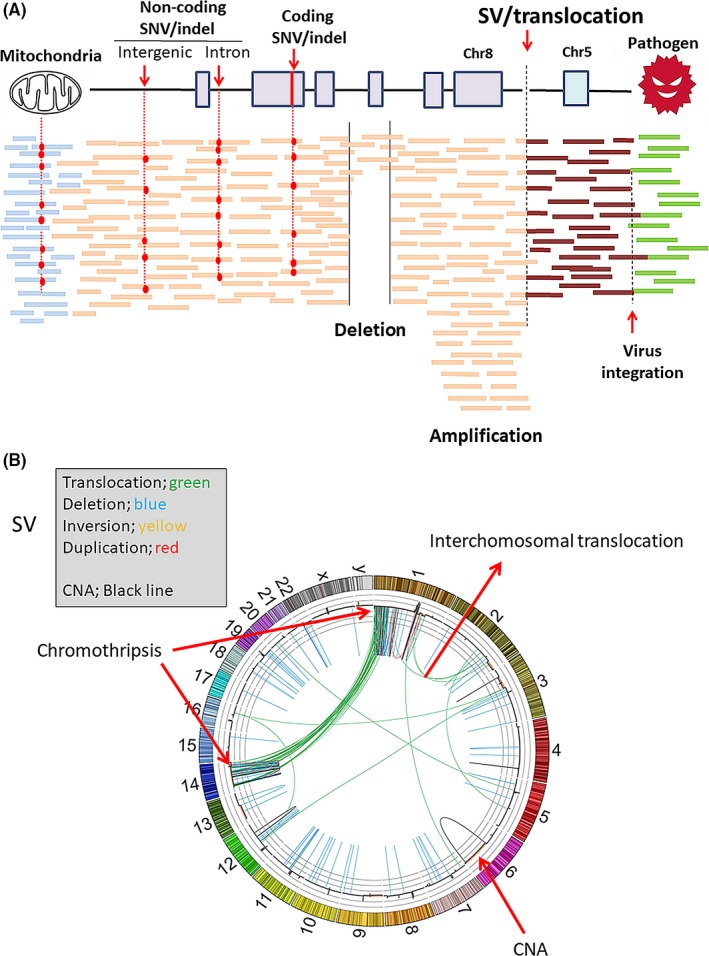
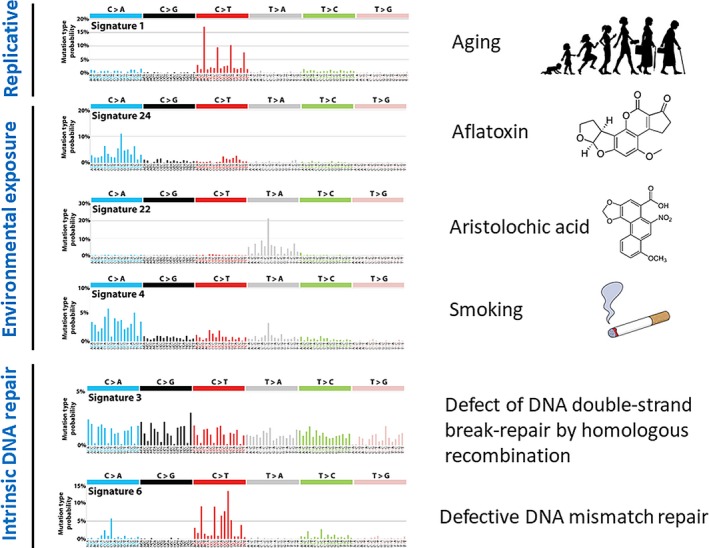
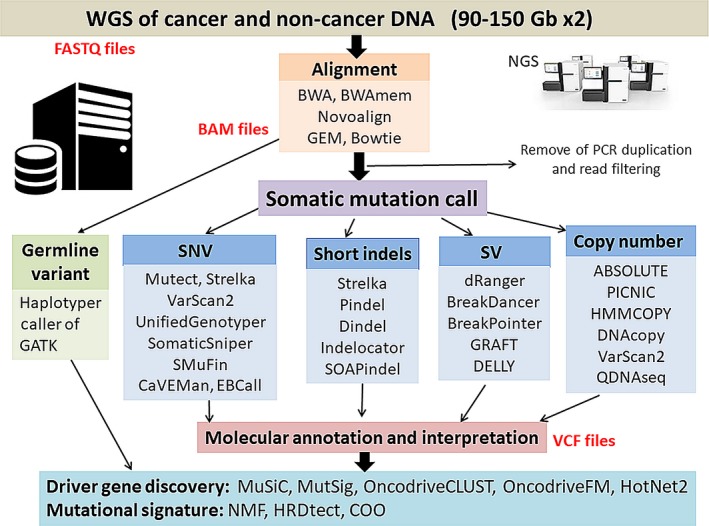
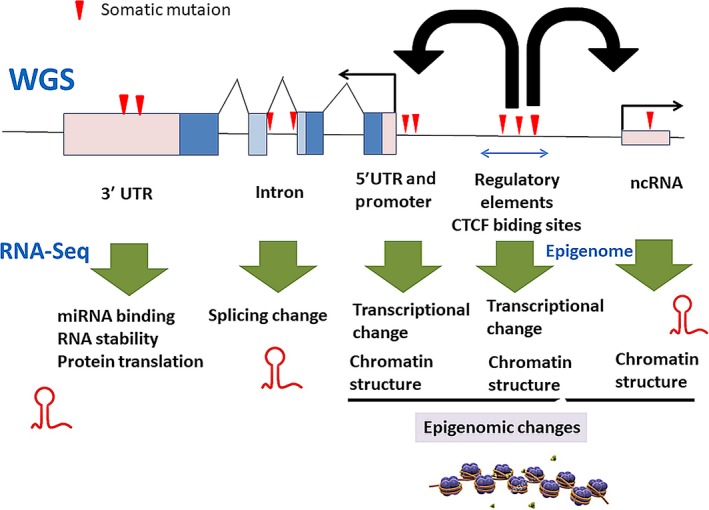
Explosive advances in next-generation sequencer (NGS) and computational analyses have enabled exploration of somatic protein-altered mutations in most cancer types, with coding mutation data intensively accumulated. However, there is limited information on somatic mutations in non-coding regions, including introns, regulatory elements and non-coding RNA. Structural variants and pathogen in cancer genomes remain widely unexplored. Whole genome sequencing (WGS) approaches can be used to comprehensively explore all types of genomic alterations in cancer and help us to better understand the whole landscape of driver mutations and mutational signatures in cancer genomes and elucidate the functional or clinical implications of these unexplored genomic regions and mutational signatures. This review describes recently developed technical approaches for cancer WGS and the future direction of cancer WGS, and discusses its utility and limitations as an analysis platform and for mutation interpretation for cancer genomics and cancer precision medicine. Taking into account the diversity of cancer genomes and phenotypes, interpretation of abundant mutation information from WGS, especially non-coding and structure variants, requires the analysis of large-scale WGS data integrated with RNA-Seq, epigenomics, immuno-genomic and clinic-pathological information.
下一代测序仪(NGS)和计算分析技术的迅猛发展,使得在大多数癌症类型中能够探索体细胞蛋白改变突变,编码突变数据大量积累。然而,关于非编码区体细胞突变的信息有限,包括内含子、调控元件和非编码RNA。癌症基因组中的结构变异和病原体仍未得到广泛探索。全基因组测序(WGS)方法可用于全面探索癌症中所有类型的基因组改变,帮助我们更好地理解癌症基因组中驱动突变和突变特征的全貌,并阐明这些未探索的基因组区域和突变特征的功能或临床意义。本文综述了近期开发的癌症WGS技术方法及其未来发展方向,并讨论了其作为癌症基因组学和癌症精准医学分析平台及突变解读的效用和局限性。考虑到癌症基因组和表型的多样性,对来自WGS的大量突变信息进行解读,尤其是非编码和结构变异,需要分析整合了RNA测序、表观基因组学、免疫基因组学和临床病理信息的大规模WGS数据。