Neonatology Unit, Department of Pediatrics, Security Forces Hospital, Riyadh, 11481, Saudi Arabia.
Division of Pediatric Neurology, Department of Pediatrics, College of Medicine, King Saud University, Riyadh, Saudi Arabia.
BMC Neurol. 2020 May 25;20(1):207. doi: 10.1186/s12883-020-01761-w.
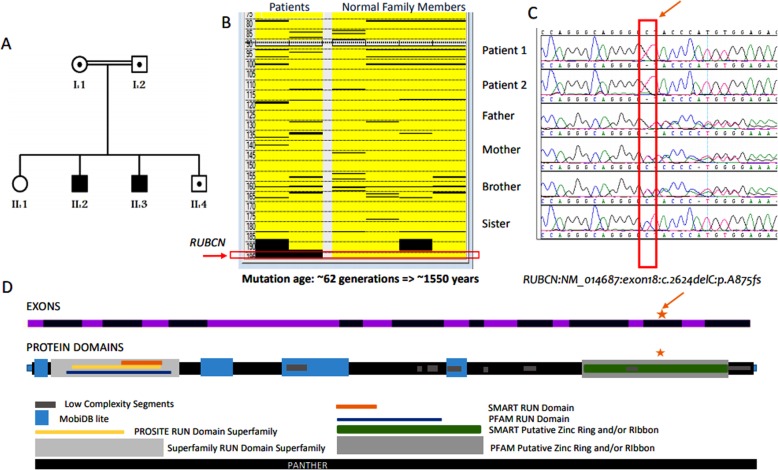
Homozygous frameshift mutation in RUBCN (KIAA0226), known to result in endolysosomal machinery defects, has previously been reported in a single Saudi family with autosomal recessive spinocerebellar ataxia (Salih ataxia, SCAR15, OMIM # 615705). The present report describes the clinical, neurophysiologic, neuroimaging, and genetic findings in a second unrelated Saudi family with two affected children harboring identical homozygous frameshift mutation in the gene. It also explores and documents an ancient founder cerebellar ataxia mutation in the Arabian Peninsula.
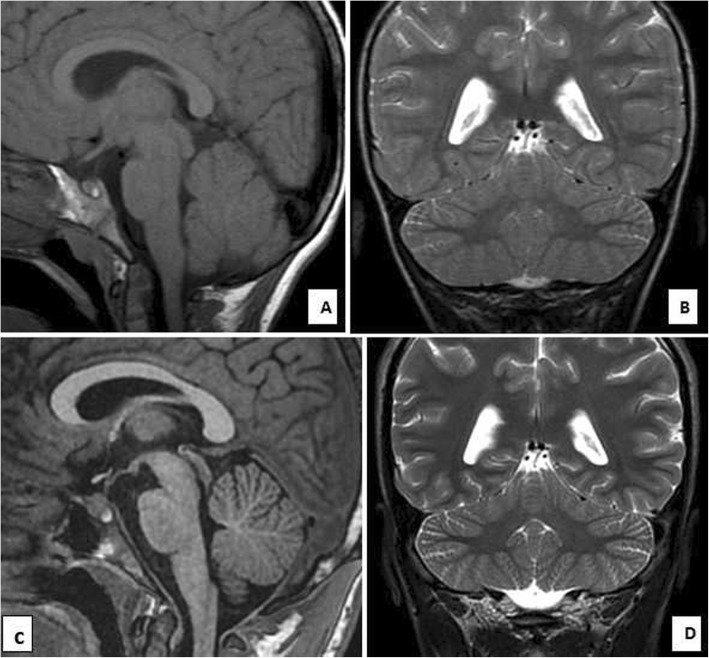
The present family has two affected males (aged 6.5 and 17 years) with unsteady gait apparent since learning to walk at 2.5 and 3 years, respectively. The younger patient showed gait ataxia and normal reflexes. The older patient had saccadic eye movement, dysarthria, mild upper and lower limb and gait ataxia (on tandem walking), and enhanced reflexes in the lower limbs. Cognitive abilities were mildly impaired in the younger sibling (IQ 67) and borderline in the older patient (IQ 72). Nerve conduction studies were normal in both patients. MRI was normal at 2.5 years in the younger sibling. Brain MRI showed normal cerebellar volume and folia in the older sibling at the age of 6 years, and revealed minimal superior vermian atrophy at the age of 16 years. Autozygome and exome analysis showed both affected have previously reported homoallelic mutation in RUBCN (NM_014687:exon18:c.2624delC:p.A875fs), whereas the parents are carriers. Autozygosity mapping focused on smallest haplotype on chromosome 3 and mutation age analysis revealed the mutation occurred approximately 1550 years ago spanning about 62 generations.
Our findings validate the slowly progressive phenotype of Salih ataxia (SCAR15, OMIM # 615705) by an additional family. Haplotype sharing attests to a common founder, an ancient RUBCN mutation in the Arab population.
已知 RUBCN(KIAA0226)中的纯合移码突变可导致内溶酶体机制缺陷,先前在一个患有常染色体隐性脊髓小脑共济失调(Salih 共济失调,SCAR15,OMIM#615705)的沙特单一家庭中报道过。本报告描述了第二个无关沙特家庭的临床、神经生理、神经影像学和遗传发现,该家庭的两个受影响的孩子携带该基因的相同纯合移码突变。它还探索并记录了阿拉伯半岛的一种古老的小脑共济失调突变。
本家系有两个受影响的男性(分别为 6.5 岁和 17 岁),分别在 2.5 岁和 3 岁学会行走时开始出现不稳定的步态。年轻患者表现为步态共济失调和正常反射。年长的患者有扫视眼运动、构音障碍、轻度上下肢和步态共济失调(在走平衡木时)以及下肢反射增强。年轻的兄弟姐妹认知能力轻度受损(智商 67),年长的患者认知能力边缘受损(智商 72)。神经传导研究在两个患者中均正常。年轻患者在 2.5 岁时的 MRI 正常。年长患者在 6 岁时的脑 MRI 显示正常小脑体积和叶片,在 16 岁时显示轻微的上蚓部萎缩。自交系和外显子组分析显示,两个受影响的患者均携带先前报道的 RUBCN 同源等位基因突变(NM_014687:exon18:c.2624delC:p.A875fs),而父母则为携带者。自交系作图集中在第 3 号染色体上的最小单倍型,突变年龄分析显示突变发生在大约 1550 年前,跨越大约 62 代。
我们的发现通过另一个家系验证了 Salih 共济失调(SCAR15,OMIM#615705)的缓慢进展表型。单倍型共享证明了阿拉伯人群中存在一个共同的 RUBCN 突变的古老祖先。