Department of Chemistry - Ångström Laboratory, Uppsala University, SE-751 20, Uppsala, Sweden.
Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo námĕstí 2, 16610 Prague 6, Czech Republic.
J Am Chem Soc. 2020 Jun 24;142(25):10942-10954. doi: 10.1021/jacs.9b13769. Epub 2020 Jun 11.
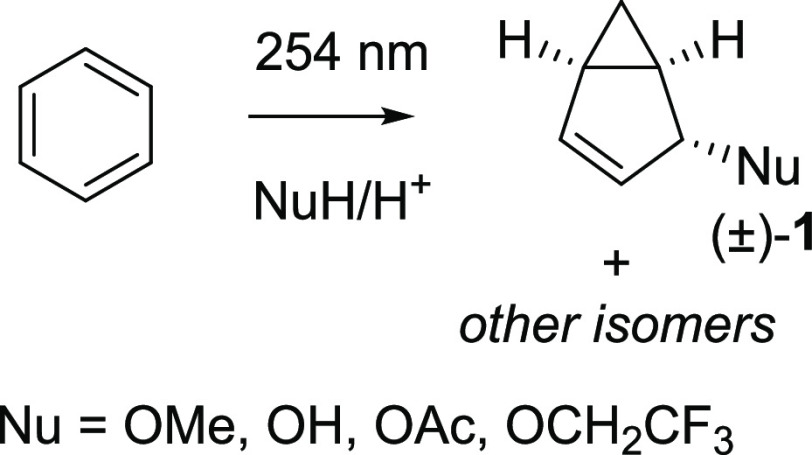
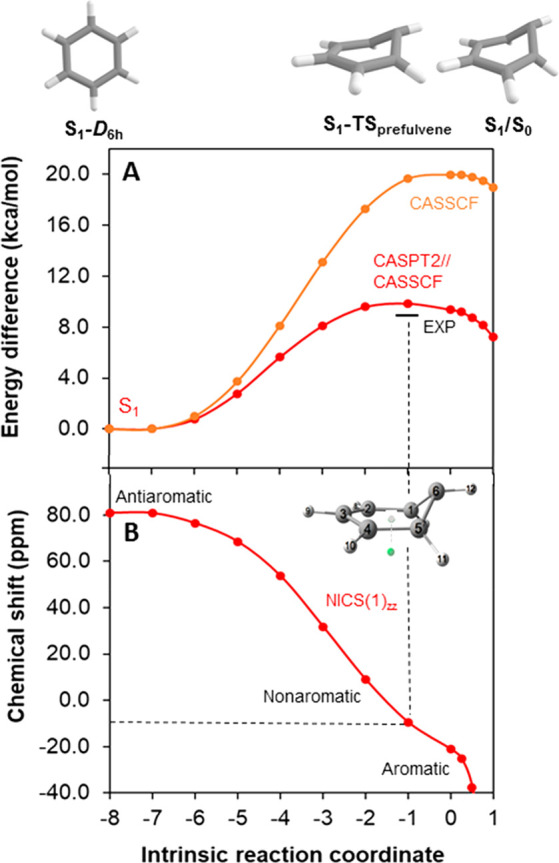
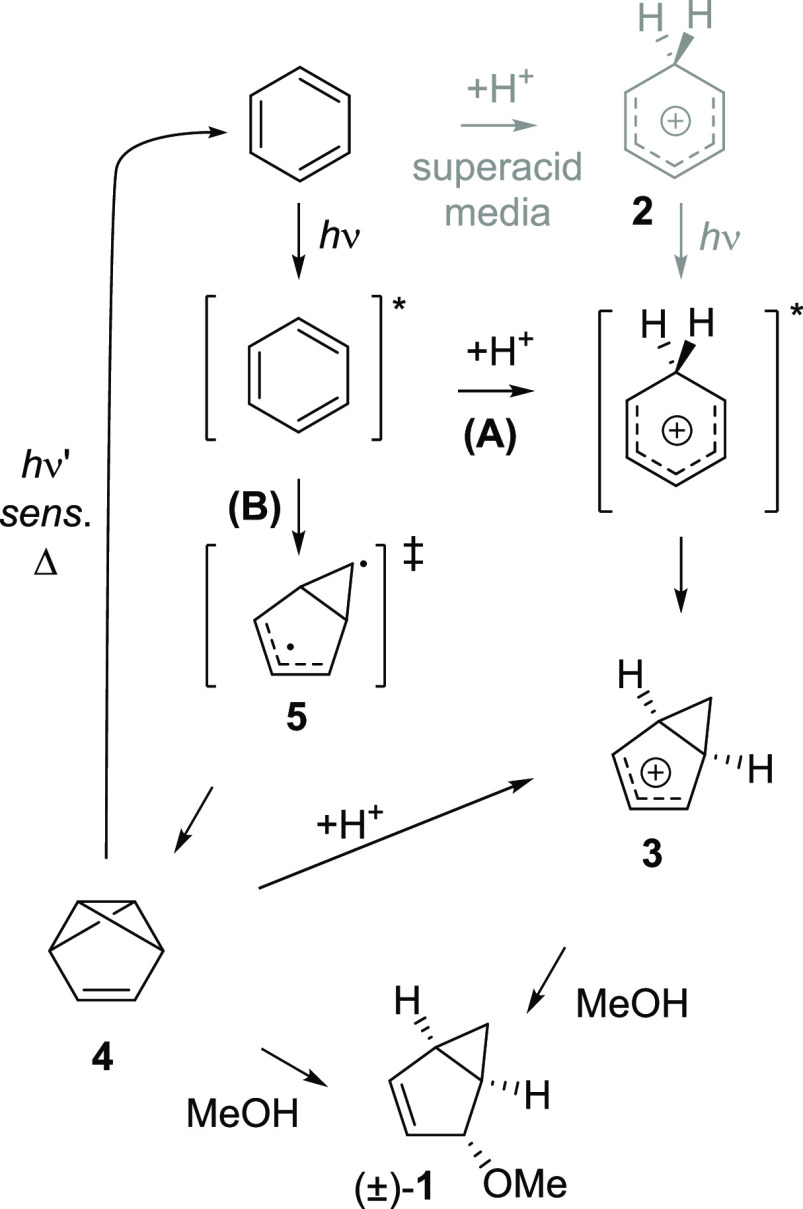
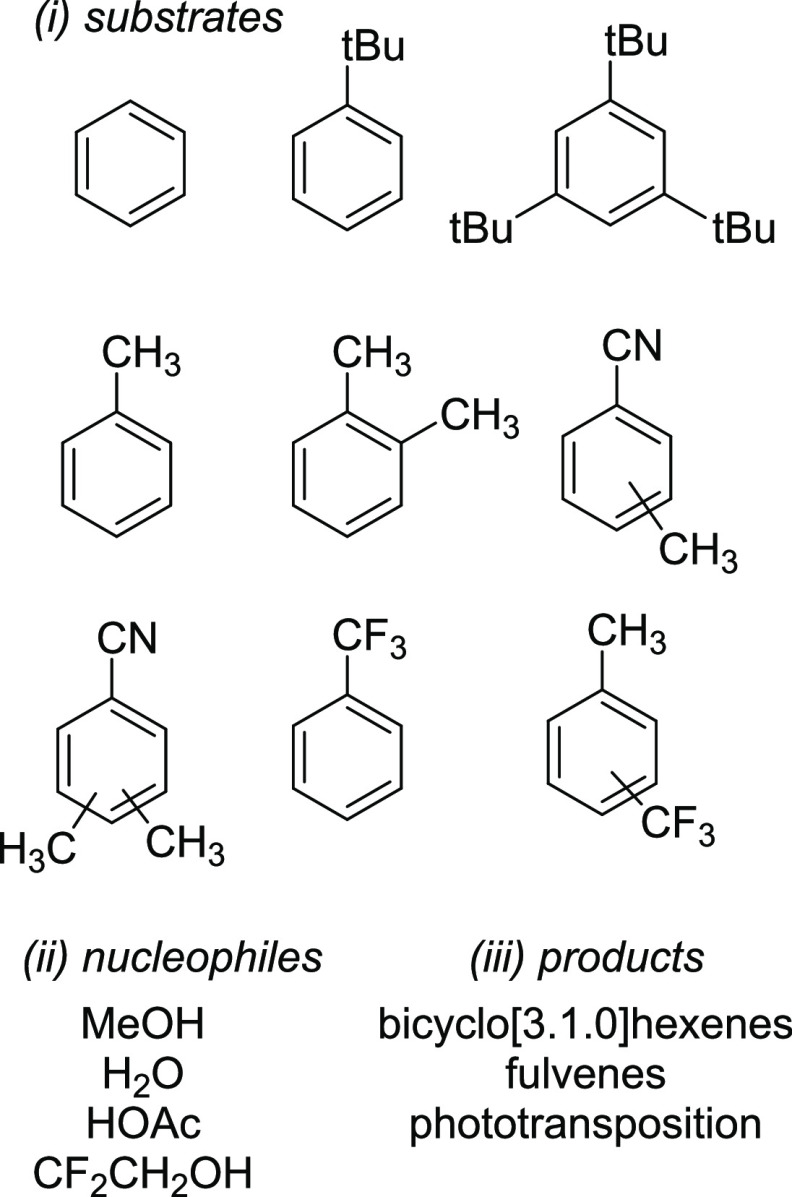
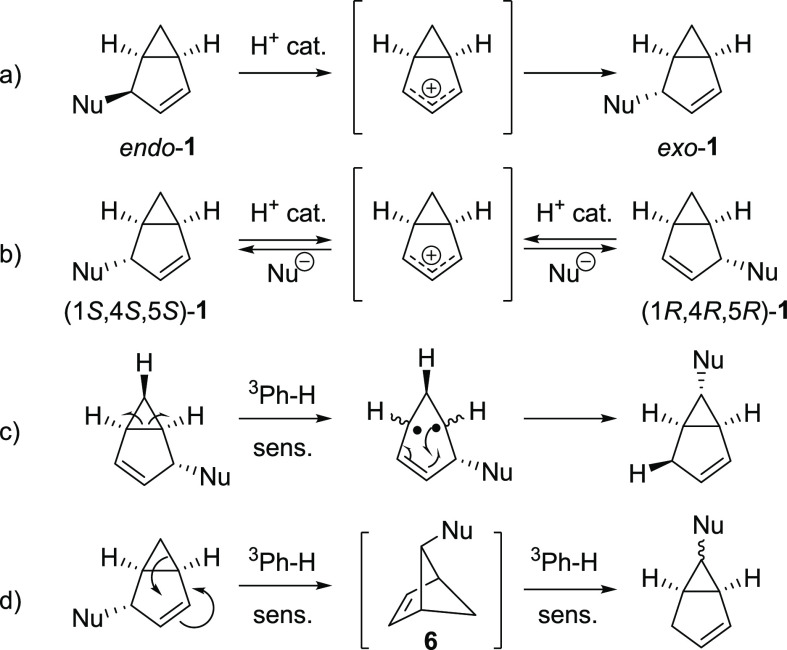
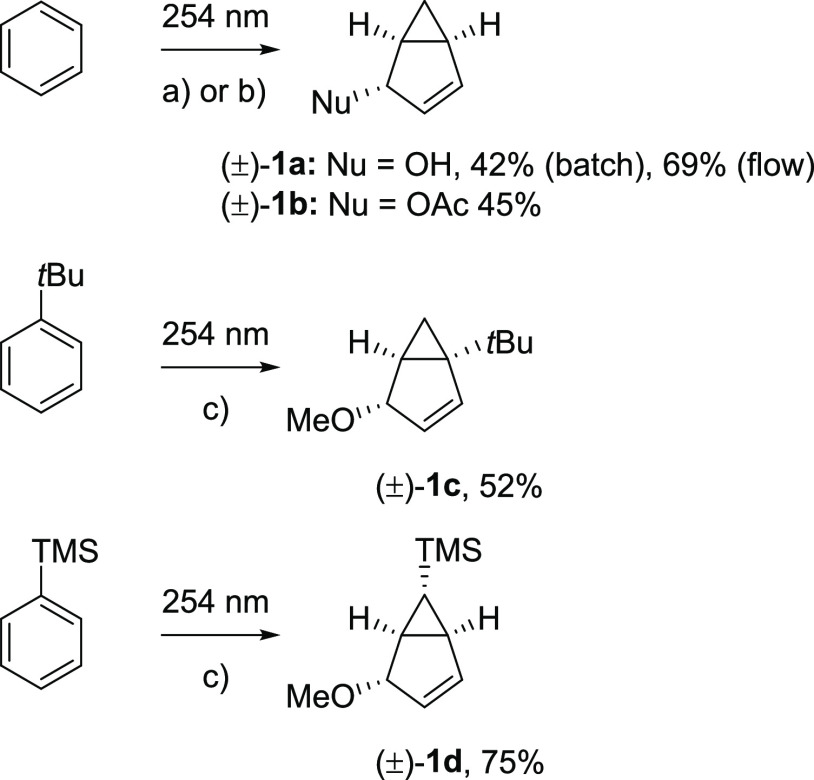
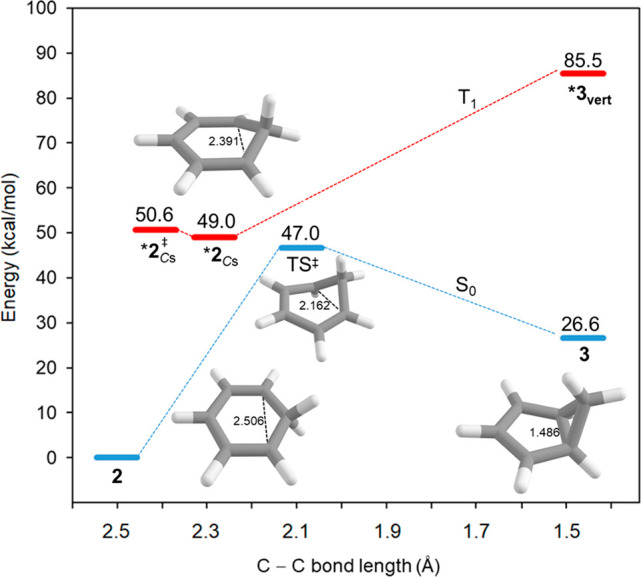
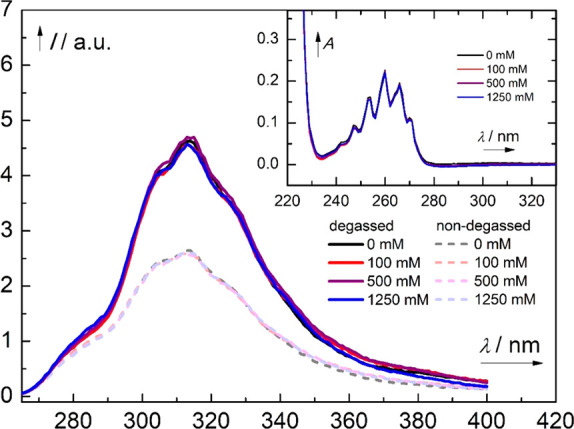
Benzene exhibits a rich photochemistry which can provide access to complex molecular scaffolds that are difficult to access with reactions in the electronic ground state. While benzene is aromatic in its ground state, it is antiaromatic in its lowest ππ* excited states. Herein, we clarify to what extent relief of excited-state antiaromaticity (ESAA) triggers a fundamental benzene photoreaction: the photoinitiated nucleophilic addition of solvent to benzene in acidic media leading to substituted bicyclo[3.1.0]hex-2-enes. The reaction scope was probed experimentally, and it was found that silyl-substituted benzenes provide the most rapid access to bicyclo[3.1.0]hexene derivatives, formed as single isomers with three stereogenic centers in yields up to 75% in one step. Two major mechanism hypotheses, both involving ESAA relief, were explored through quantum chemical calculations and experiments. The first mechanism involves protonation of excited-state benzene and subsequent rearrangement to bicyclo[3.1.0]hexenium cation, trapped by a nucleophile, while the second involves photorearrangement of benzene to benzvalene followed by protonation and nucleophilic addition. Our studies reveal that the second mechanism is operative. We also clarify that similar ESAA relief leads to puckering of S-state silabenzene and pyridinium ion, where the photorearrangement of the latter is of established synthetic utility. Finally, we identified causes for the limitations of the reaction, information that should be valuable in explorations of similar photoreactions. Taken together, we reveal how the ESAA in benzene and 6π-electron heterocycles trigger photochemical distortions that provide access to complex three-dimensional molecular scaffolds from simple reactants.
苯具有丰富的光化学反应性,可以提供难以通过电子基态反应获得的复杂分子骨架。虽然苯在基态下是芳香的,但在其最低的ππ*激发态下是反芳香的。在此,我们澄清了在多大程度上缓解激发态反芳香性(ESAA)引发了基本的苯光反应:在酸性介质中溶剂对苯的光引发亲核加成,导致取代的双环[3.1.0]己-2-烯。通过实验探测了反应范围,发现取代的苯提供了最快的途径来获得双环[3.1.0]己烯衍生物,以三种立体中心的单一异构体形式形成,产率高达 75%,一步完成。通过量子化学计算和实验探索了两种主要的机制假设,都涉及 ESAA 缓解。第一种机制涉及激发态苯的质子化,随后重排为双环[3.1.0]己烯阳离子,被亲核试剂捕获,而第二种机制涉及苯到苯并戊二烯的光重排,随后质子化和亲核加成。我们的研究表明第二种机制是可行的。我们还澄清了类似的 ESAA 缓解导致 S-态硅苯和吡啶鎓离子的卷曲,其中后者的光重排具有既定的合成用途。最后,我们确定了反应限制的原因,这些信息在探索类似的光反应时应该是有价值的。综上所述,我们揭示了苯和 6π 电子杂环中的 ESAA 如何引发光化学扭曲,从而从简单的反应物获得复杂的三维分子骨架。