Frid Maria G, Thurman Joshua M, Hansen Kirk C, Maron Bradley A, Stenmark Kurt R
University of Colorado, Denver, Anschutz Medical Campus, USA.
Brigham and Women's Hospital, Harvard Medical School, USA.
Glob Cardiol Sci Pract. 2020 Apr 30;2020(1):e202001. doi: 10.21542/gcsp.2020.1.
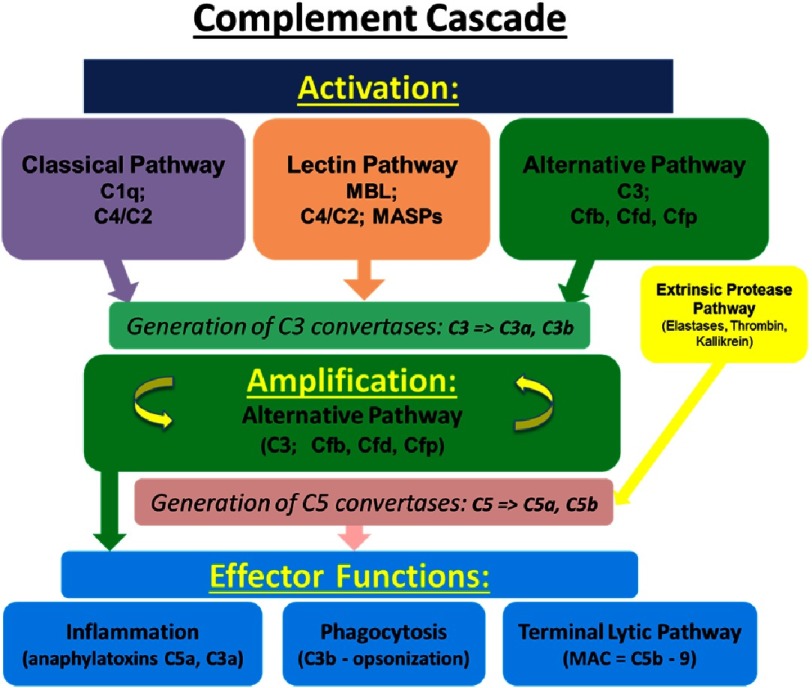
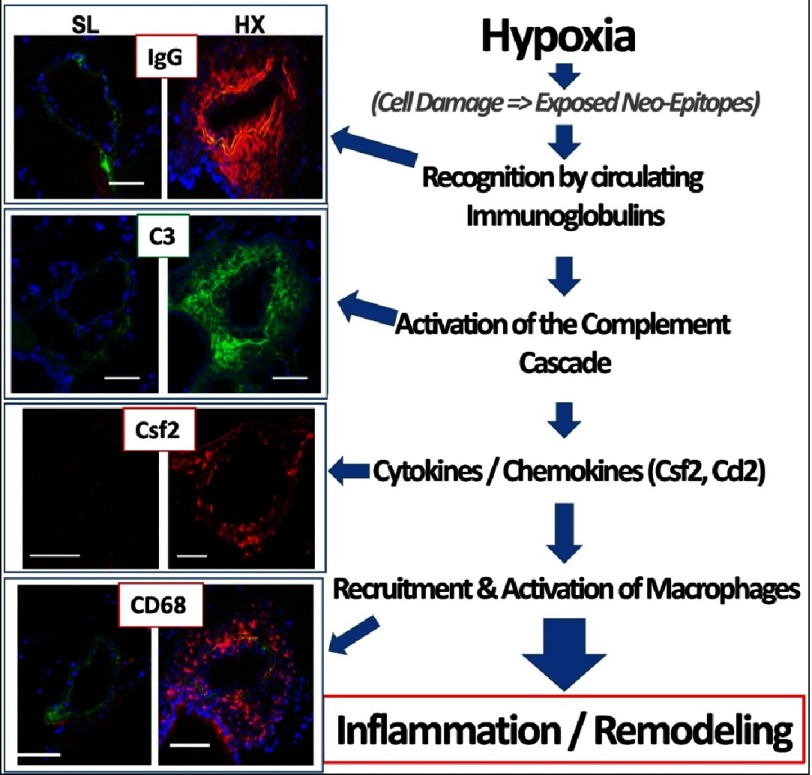
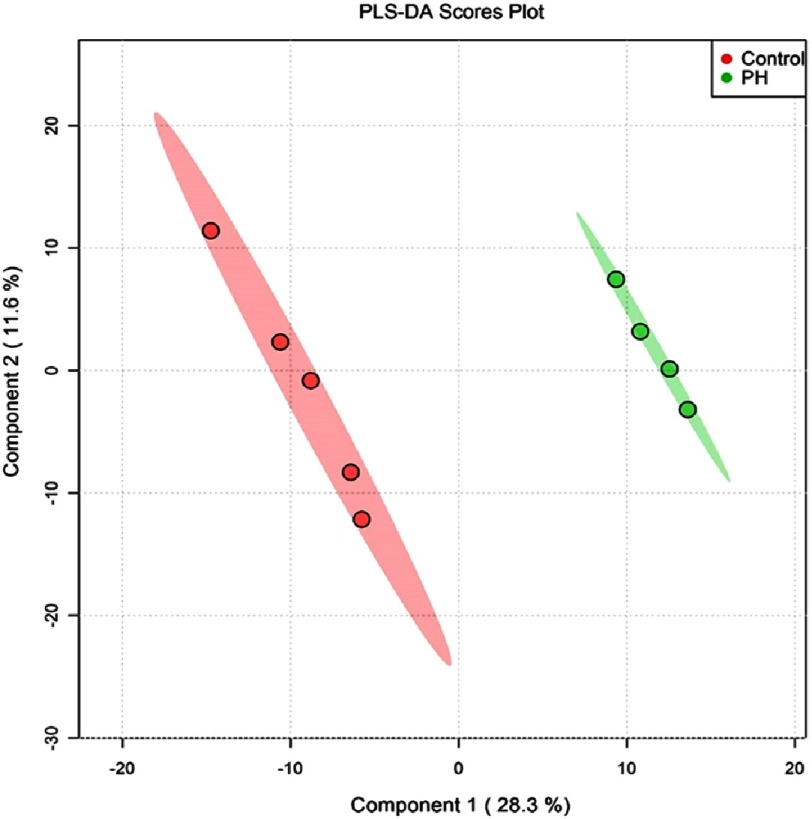
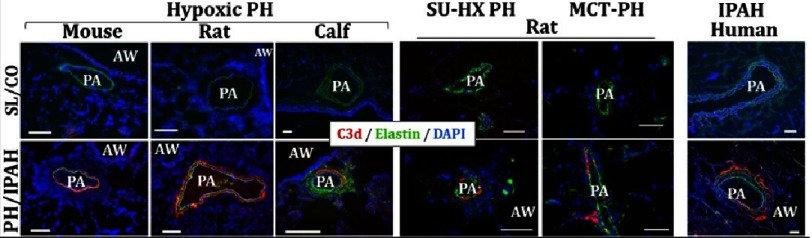
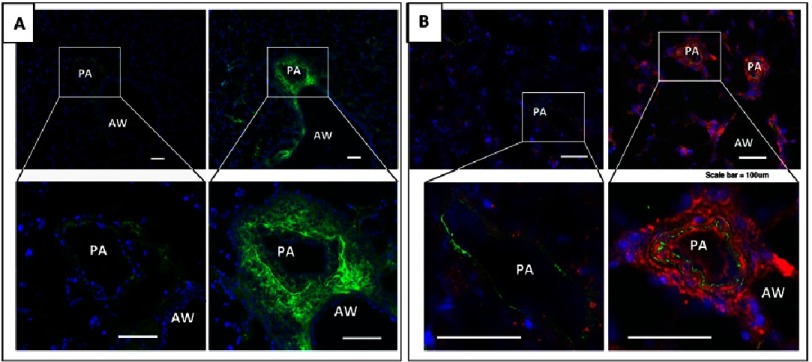
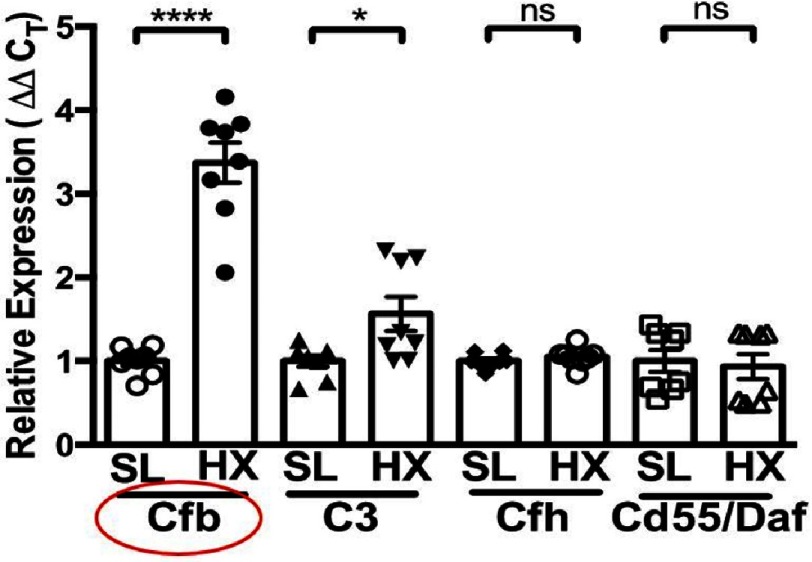
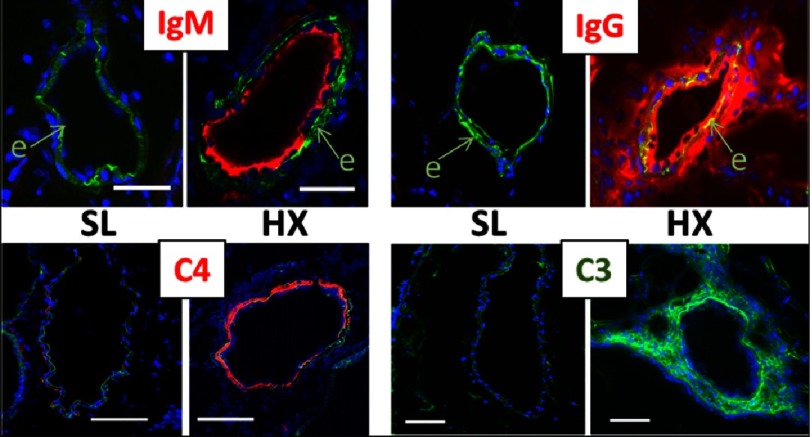
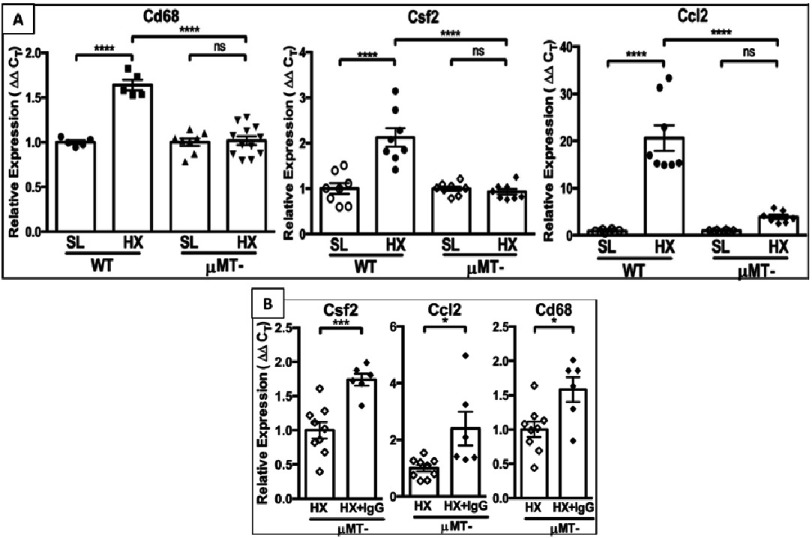
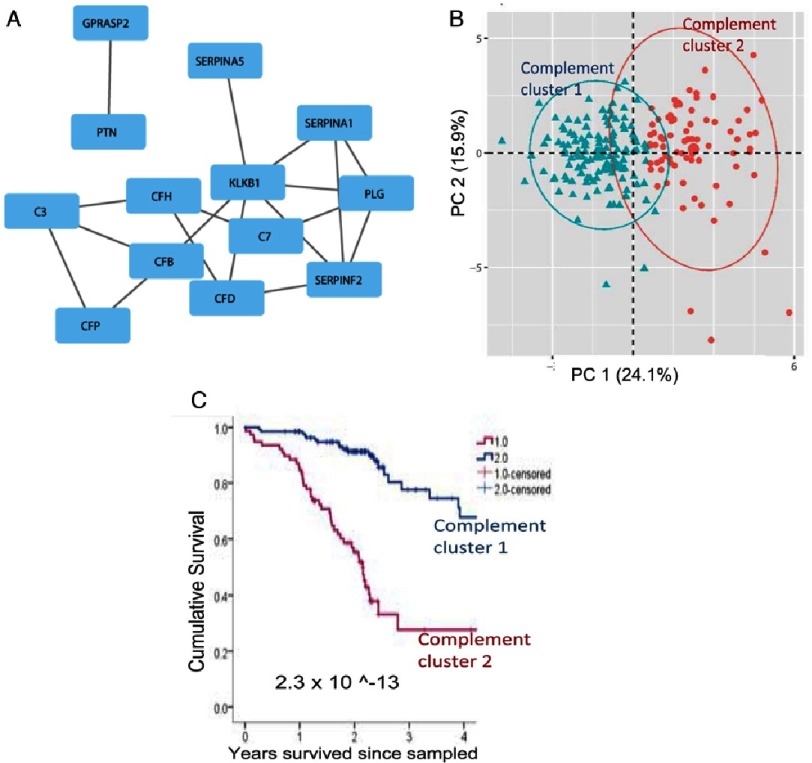
Pulmonary (arterial) hypertension (PH/PAH) is a life-threatening cardiopulmonary disorder. Experimental evidence suggests involvement of inflammatory and autoimmune processes in pathogenesis of PH/PAH, however the triggering and disease-promoting mechanisms remain unknown. The complement system is a key arm of innate immunity implicated in various pro-inflammatory and autoimmune diseases, yet, surprisingly little is known about the role of complement in PH/PAH pathogenesis. The preponderance of the existing data associates complement with PH/PAH via analysis of plasma and does not study the lung directly. Therefore, we aimed to resolve this by analyzing both the mechanisms of local lung-specific complement activation and the correlation of dysregulated plasma complement to clinical outcome in PAH patients. In our recent studies, reviewed herein, we show, for the first time, that immunoglobulin-driven activation of the complement cascade, specifically its alternative pathway, in the pulmonary perivascular areas, is a key mechanism initiating pro-inflammatory processes in the early stage of experimental hypoxic PH (a form of "sterile inflammation"). In human patients with end-stage PAH, we have demonstrated that perivascular deposition of immunoglobulin G (IgG) and activation of the complement cascade are "longitudinally" persistent in the disease. We also showed, using unbiased network analysis, that plasma complement signaling, including again the Alternative pathway, is a prognostic factor of survival in patients with idiopathic PAH (IPAH). Based on these initial findings, we suggest that vascular-specific, immunoglobulin-driven dysregulated complement signaling triggers and maintains pulmonary vascular remodeling and PH. Future experiments in this area would facilitate discoveries on whether complement signaling can serve both as a biomarker and therapeutic target in PH/PAH.
肺动脉高压(PH/PAH)是一种危及生命的心肺疾病。实验证据表明,炎症和自身免疫过程参与了PH/PAH的发病机制,然而,引发和促进疾病的机制仍不清楚。补体系统是固有免疫的关键组成部分,与多种促炎和自身免疫性疾病有关,然而,令人惊讶的是,关于补体在PH/PAH发病机制中的作用知之甚少。现有数据大多通过血浆分析将补体与PH/PAH联系起来,而没有直接研究肺。因此,我们旨在通过分析局部肺特异性补体激活机制以及血浆补体失调与PAH患者临床结局的相关性来解决这一问题。在本文回顾的我们最近的研究中,我们首次表明,免疫球蛋白驱动的补体级联激活,特别是其替代途径,在肺血管周围区域,是实验性低氧性PH(一种“无菌性炎症”形式)早期启动促炎过程的关键机制。在终末期PAH的人类患者中,我们已经证明免疫球蛋白G(IgG)的血管周围沉积和补体级联激活在疾病中是“纵向”持续存在的。我们还使用无偏网络分析表明,血浆补体信号传导,同样包括替代途径,是特发性PAH(IPAH)患者生存的预后因素。基于这些初步发现,我们认为血管特异性、免疫球蛋白驱动的补体信号失调触发并维持肺血管重塑和PH。该领域未来的实验将有助于发现补体信号是否可以作为PH/PAH的生物标志物和治疗靶点。