Department of Ecology and Evolutionary Biology, University of California Los Angeles, California 90095-7246.
Department of Biology, Stanford University, Stanford, California 94305.
Genetics. 2020 Jul;215(3):799-812. doi: 10.1534/genetics.120.303081. Epub 2020 Jun 2.
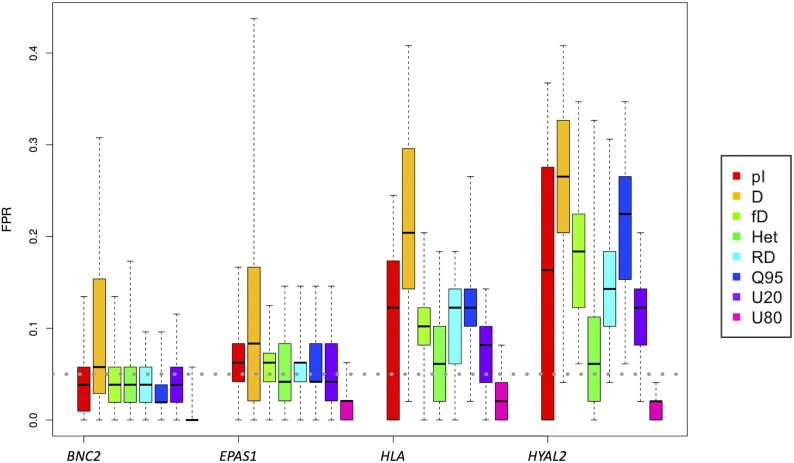
Admixture with archaic hominins has altered the landscape of genomic variation in modern human populations. Several gene regions have been identified previously as candidates of adaptive introgression (AI) that facilitated human adaptation to specific environments. However, simulation-based studies have suggested that population genetic processes other than adaptive mutations, such as heterosis from recessive deleterious variants private to populations before admixture, can also lead to patterns in genomic data that resemble AI. The extent to which the presence of deleterious variants affect the false-positive rate and the power of current methods to detect AI has not been fully assessed. Here, we used extensive simulations under parameters relevant for human evolution to show that recessive deleterious mutations can increase the false positive rates of tests for AI compared to models without deleterious variants, especially when the recombination rates are low. We next examined candidates of AI in modern humans identified from previous studies, and show that 24 out of 26 candidate regions remain significant, even when deleterious variants are included in the null model. However, two AI candidate genes, and , are particularly susceptible to high false positive signals of AI due to recessive deleterious mutations. These genes are located in regions of the human genome with high exon density together with low recombination rate, factors that we show increase the rate of false-positives due to recessive deleterious mutations. Although the combination of such parameters is rare in the human genome, caution is warranted in such regions, as well as in other species with more compact genomes and/or lower recombination rates. In sum, our results suggest that recessive deleterious mutations cannot account for the signals of AI in most, but not all, of the top candidates for AI in humans, suggesting they may be genuine signals of adaptation.
古人类基因混合改变了现代人类群体基因组变异的格局。先前已经确定了几个基因区域作为适应性渗入(AI)的候选者,这些基因有助于人类适应特定环境。然而,基于模拟的研究表明,除了适应性突变之外的种群遗传过程,例如在混合之前种群中特有的隐性有害变异的杂种优势,也可以导致基因组数据中出现类似于 AI 的模式。有害变异的存在对假阳性率和当前检测 AI 方法的功效的影响程度尚未得到充分评估。在这里,我们使用与人类进化相关的参数进行了广泛的模拟,结果表明隐性有害突变会增加 AI 检测的假阳性率,与没有有害变异的模型相比,尤其是在重组率较低的情况下。我们接下来检查了以前研究中鉴定的现代人类 AI 候选者,并表明即使在包含有害变异的零模型中,26 个候选区域中的 24 个仍然具有显著意义。然而,两个 AI 候选基因 和 由于隐性有害突变,特别容易受到 AI 的高假阳性信号的影响。这些基因位于人类基因组中具有高外显子密度和低重组率的区域,我们的研究表明,这些因素会增加由于隐性有害突变导致的假阳性率。尽管在人类基因组中这种参数的组合很少见,但在这些区域以及其他基因组更紧凑和/或重组率更低的物种中,都需要谨慎对待。总之,我们的结果表明,在人类 AI 的大多数顶级候选者中,隐性有害突变不能解释 AI 的信号,但在某些情况下,它们可能是真正的适应信号。