Hanke Nina, Türk Denise, Selzer Dominik, Wiebe Sabrina, Fernandez Éric, Stopfer Peter, Nock Valerie, Lehr Thorsten
Clinical Pharmacy, Saarland University, 66123 Saarbrücken, Germany.
Translational Medicine and Clinical Pharmacology, Boehringer Ingelheim Pharma GmbH & Co. KG, 88397 Biberach, Germany.
Pharmaceutics. 2020 Jun 16;12(6):556. doi: 10.3390/pharmaceutics12060556.
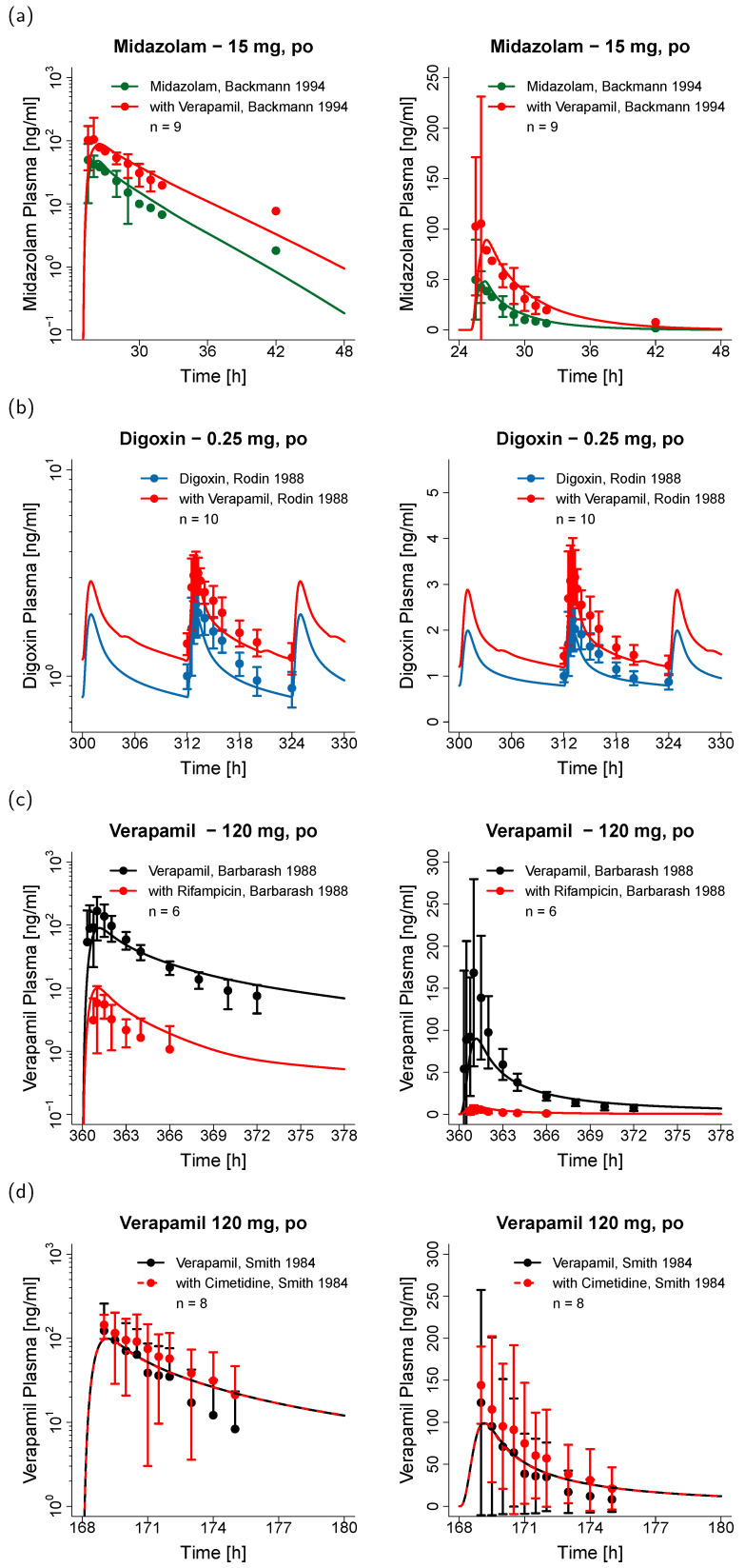
The calcium channel blocker and antiarrhythmic agent verapamil is recommended by the FDA for drug-drug interaction (DDI) studies as a moderate clinical CYP3A4 index inhibitor and as a clinical Pgp inhibitor. The purpose of the presented work was to develop a mechanistic whole-body physiologically based pharmacokinetic (PBPK) model to investigate and predict DDIs with verapamil. The model was established in PK-Sim, using 45 clinical studies (dosing range 0.1-250 mg), including literature as well as unpublished Boehringer Ingelheim data. The verapamil R- and S-enantiomers and their main metabolites R- and S-norverapamil are represented in the model. The processes implemented to describe the pharmacokinetics of verapamil and norverapamil include enantioselective plasma protein binding, enantioselective metabolism by CYP3A4, non-stereospecific Pgp transport, and passive glomerular filtration. To describe the auto-inhibitory and DDI potential, mechanism-based inactivation of CYP3A4 and non-competitive inhibition of Pgp by the verapamil and norverapamil enantiomers were incorporated based on in vitro literature. The resulting DDI performance was demonstrated by prediction of DDIs with midazolam, digoxin, rifampicin, and cimetidine, with 21/22 predicted DDI AUC ratios or C ratios within 1.5-fold of the observed values. The thoroughly built and qualified model will be freely available in the Open Systems Pharmacology model repository to support model-informed drug discovery and development.
钙通道阻滞剂及抗心律失常药维拉帕米被美国食品药品监督管理局(FDA)推荐用于药物相互作用(DDI)研究,作为中度临床CYP3A4指标抑制剂及临床P-糖蛋白(Pgp)抑制剂。本研究的目的是建立一个基于生理药代动力学(PBPK)的全身机制模型,以研究和预测与维拉帕米的药物相互作用。该模型在PK-Sim中建立,使用了45项临床研究(给药范围0.1 - 250 mg),包括文献资料以及勃林格殷格翰未发表的数据。模型中纳入了维拉帕米的R型和S型对映体及其主要代谢产物R型和S型去甲维拉帕米。描述维拉帕米和去甲维拉帕米药代动力学的过程包括对映体选择性血浆蛋白结合、CYP3A4介导的对映体选择性代谢、非立体特异性Pgp转运以及被动肾小球滤过。为描述自身抑制和药物相互作用潜力,基于体外文献,纳入了维拉帕米和去甲维拉帕米对映体对CYP3A4的基于机制的失活作用以及对Pgp的非竞争性抑制作用。通过预测与咪达唑仑、地高辛、利福平及西咪替丁之间的药物相互作用,验证了所得药物相互作用模型的性能,预测的22个药物相互作用AUC比值或C比值中有21个在观测值的1.5倍范围内。这个构建完善且经过验证的模型将在开放系统药理学模型库中免费提供,以支持基于模型的药物研发。